Department of Endocrinology Diabetology, University Hospital Center of Reims, Reims, France.
Inserm/CNRS UMR 1283/8199, Pasteur Institute of Lille, EGID, Lille, France.
Orphanet J Rare Dis. 2022 Feb 28;17(1):86. doi: 10.1186/s13023-022-02248-2.
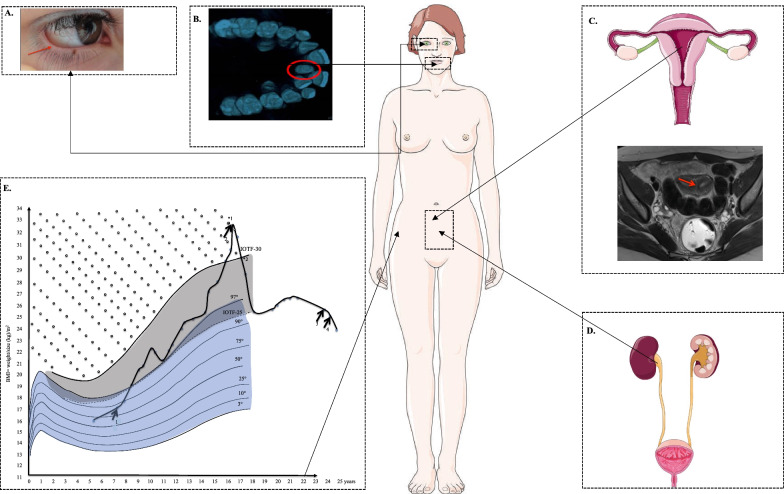
We studied a young woman with atypical diabetes associated with mild intellectual disability, lymphedema distichiasis syndrome (LDS) and polymalformative syndrome including distichiasis. We used different genetic tools to identify causative pathogenic mutations and/or copy number variations.
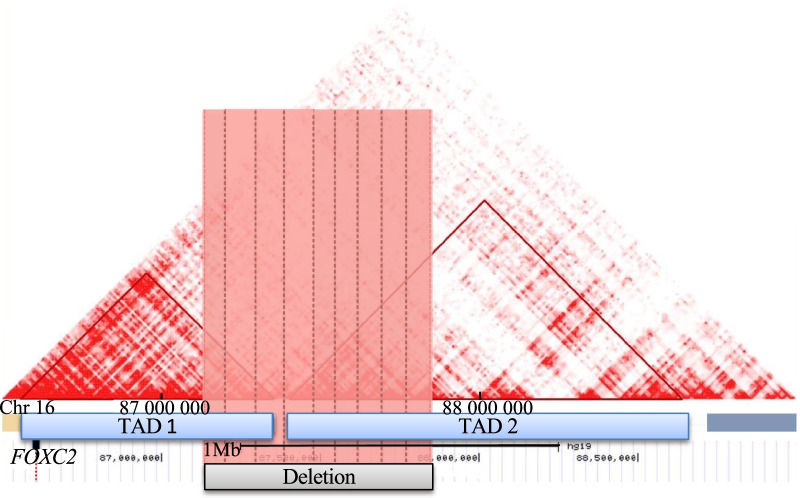
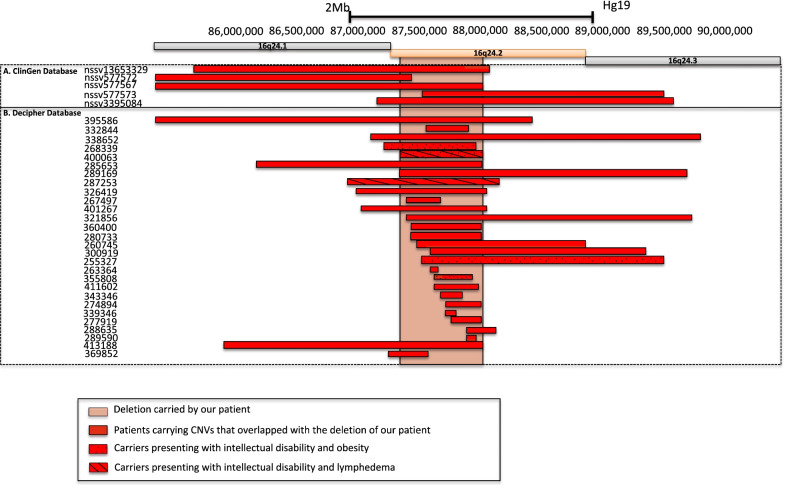
Although proband's, diabetes mellitus occurred during childhood, type 1 diabetes was unlikely due to the absence of detectable autoimmunity. DNA microarray analysis first identified a de novo, heterozygous deletion at the chr16q24.2 locus. Previously, thirty-three pathogenic or likely pathogenic deletions encompassing this locus have been reported in patients presenting with intellectual deficiency, obesity and/or lymphedema but not with diabetes. Of note, the deletion encompassed two topological association domains, whose one included FOXC2 that is known to be linked with LDS. Via whole-exome sequencing, we found a heterozygous, likely pathogenic variant in WFS1 (encoding wolframin endoplasmic reticulum [ER] transmembrane glycoprotein) which was inherited from her father who also had diabetes. WFS1 is known to be involved in monogenic diabetes. We also found a likely pathogenic variant in USP9X (encoding ubiquitin specific peptidase 9 X-linked) that is involved in X-linked intellectual disability, which was inherited from her mother who had dyscalculia and dyspraxia.
Our comprehensive genetic analysis suggested that the peculiar phenotypes of our patient were possibly due to the combination of multiple genetic causes including chr16q24.2 deletion, and two likely pathogenic variants in WFS1 and USP9X.
我们研究了一位年轻女性,她患有非典型糖尿病,伴有轻度智力障碍、淋巴管瘤多毛症(LDS)和多形性综合征,包括多毛症。我们使用不同的遗传工具来识别致病的突变和/或拷贝数变异。
虽然先证者的糖尿病发生在儿童期,但由于未检测到自身免疫,不太可能是 1 型糖尿病。DNA 微阵列分析首先在 chr16q24.2 基因座上发现了一个从头开始的杂合缺失。以前,已有 33 个致病性或可能致病性的缺失涵盖了该基因座,这些缺失与智力低下、肥胖和/或淋巴水肿有关,但与糖尿病无关。值得注意的是,该缺失涵盖了两个拓扑关联结构域,其中一个包括已知与 LDS 相关的 FOXC2。通过全外显子测序,我们发现了 WFS1(编码沃尔弗林内质网 [ER]跨膜糖蛋白)中的一个杂合的、可能致病的变异,该变异是从也患有糖尿病的父亲那里遗传来的。WFS1 已知与单基因糖尿病有关。我们还发现了 USP9X(编码泛素特异性肽酶 9 X 连锁)中的一个可能致病的变异,该变异与 X 连锁智力障碍有关,是从患有计算障碍和运动障碍的母亲那里遗传来的。
我们的综合遗传分析表明,我们患者的特殊表型可能是由于多个遗传原因的组合,包括 chr16q24.2 缺失,以及 WFS1 和 USP9X 中的两个可能致病的变异。