School of Mathematical Sciences, Peking University, Beijing, 100871, China.
Academy for Advanced Interdisciplinary Studies, Peking University, Beijing, 100871, China.
Nat Commun. 2022 Feb 28;13(1):1084. doi: 10.1038/s41467-022-28661-6.
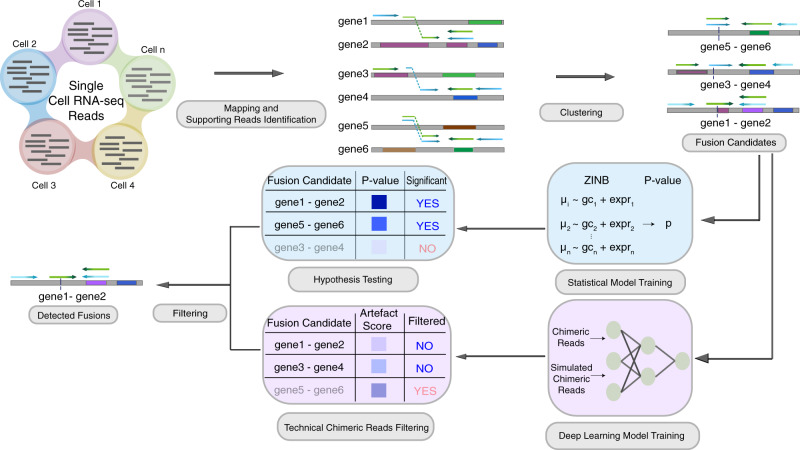
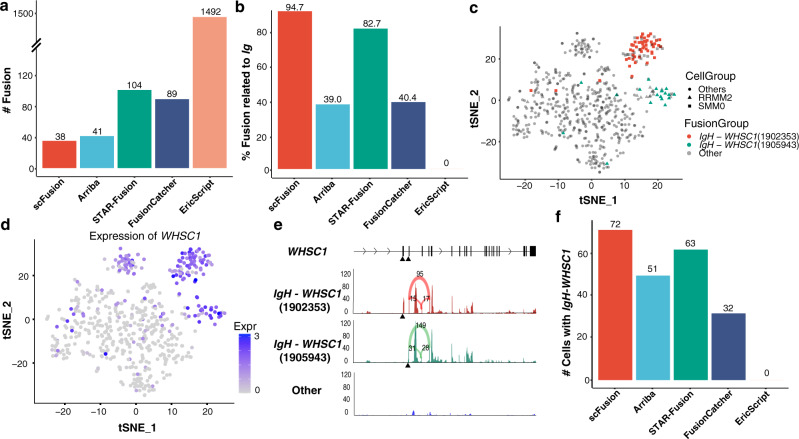
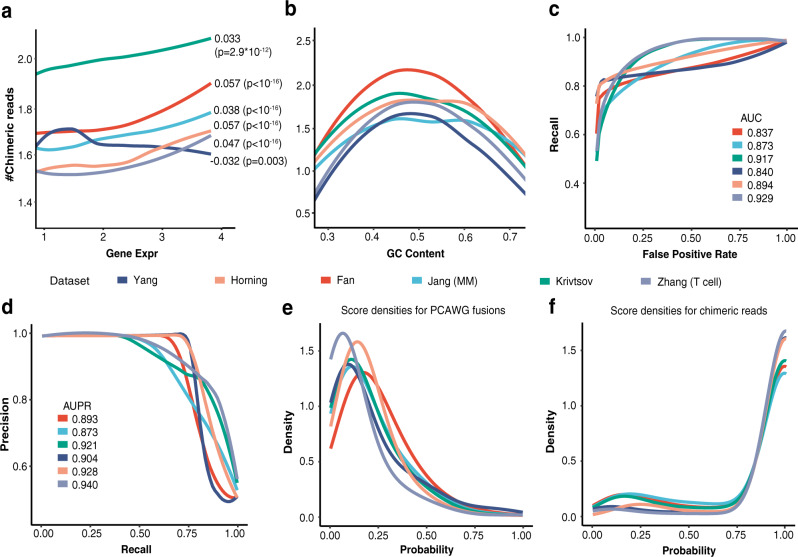
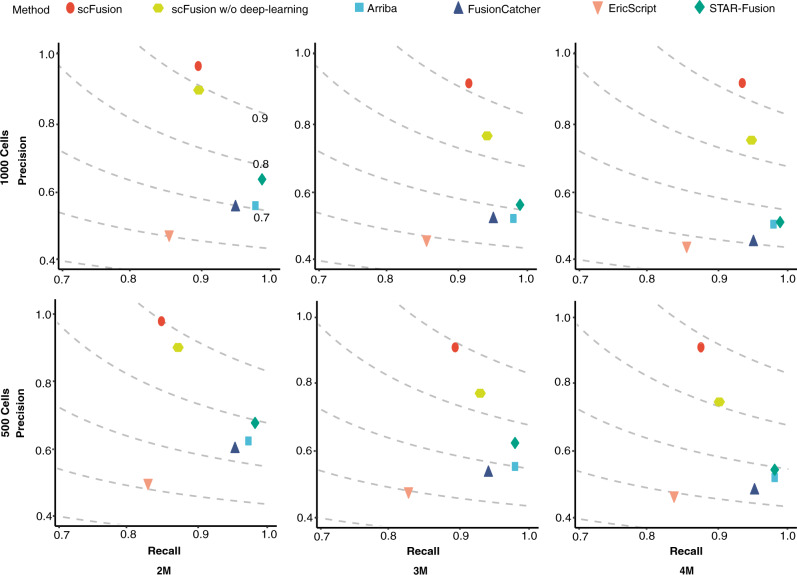
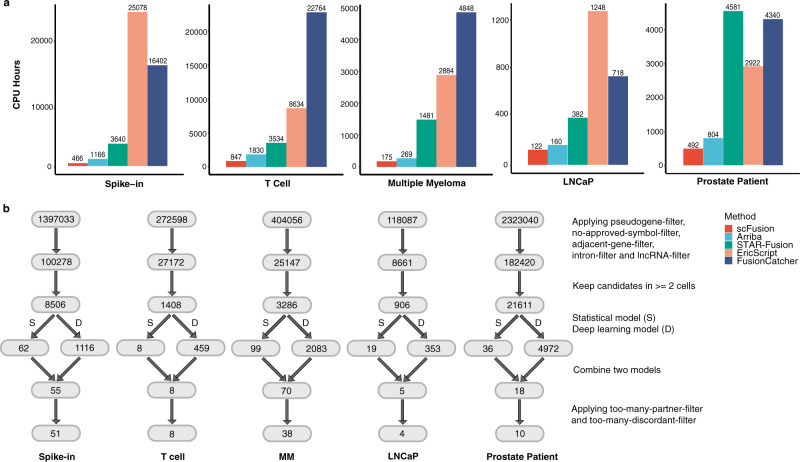
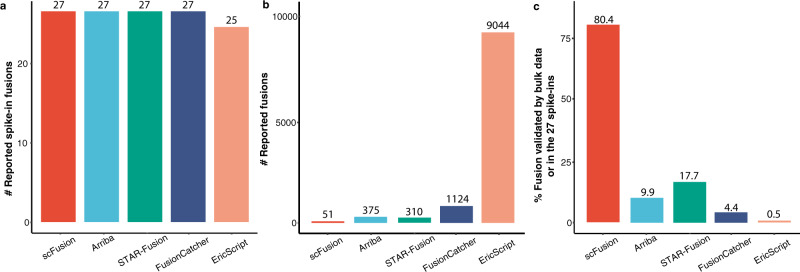
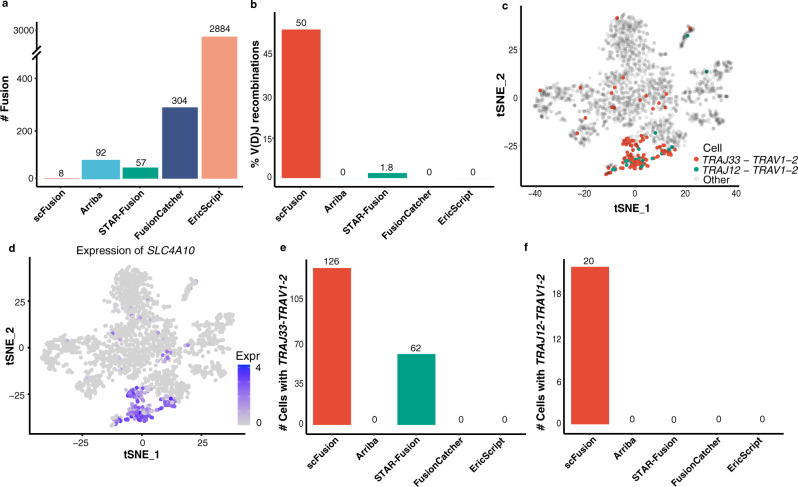
Gene fusions can play important roles in tumor initiation and progression. While fusion detection so far has been from bulk samples, full-length single-cell RNA sequencing (scRNA-seq) offers the possibility of detecting gene fusions at the single-cell level. However, scRNA-seq data have a high noise level and contain various technical artifacts that can lead to spurious fusion discoveries. Here, we present a computational tool, scFusion, for gene fusion detection based on scRNA-seq. We evaluate the performance of scFusion using simulated and five real scRNA-seq datasets and find that scFusion can efficiently and sensitively detect fusions with a low false discovery rate. In a T cell dataset, scFusion detects the invariant TCR gene recombinations in mucosal-associated invariant T cells that many methods developed for bulk data fail to detect; in a multiple myeloma dataset, scFusion detects the known recurrent fusion IgH-WHSC1, which is associated with overexpression of the WHSC1 oncogene. Our results demonstrate that scFusion can be used to investigate cellular heterogeneity of gene fusions and their transcriptional impact at the single-cell level.
基因融合在肿瘤的发生和发展中起着重要作用。虽然融合检测迄今为止一直来自于大量样本,但全长单细胞 RNA 测序 (scRNA-seq) 提供了在单细胞水平检测基因融合的可能性。然而,scRNA-seq 数据具有较高的噪声水平,并包含各种可能导致虚假融合发现的技术伪影。在这里,我们提出了一种基于 scRNA-seq 的基因融合检测的计算工具 scFusion。我们使用模拟和五个真实的 scRNA-seq 数据集来评估 scFusion 的性能,发现 scFusion 可以以低的假阳性率有效地、灵敏地检测融合。在 T 细胞数据集上,scFusion 检测到粘膜相关不变 T 细胞中的不变 TCR 基因重排,而许多为大量数据开发的方法都无法检测到这一点;在多发性骨髓瘤数据集上,scFusion 检测到已知的复发性融合 IgH-WHSC1,该融合与 WHSC1 癌基因的过表达有关。我们的结果表明,scFusion 可用于研究基因融合的细胞异质性及其在单细胞水平上的转录影响。