Nantes Université, CNRS, INSERM, l'institut du thorax, Nantes, France.
Univ Angers, SFR ICAT, Angers, France.
Front Endocrinol (Lausanne). 2022 Feb 18;13:785819. doi: 10.3389/fendo.2022.785819. eCollection 2022.
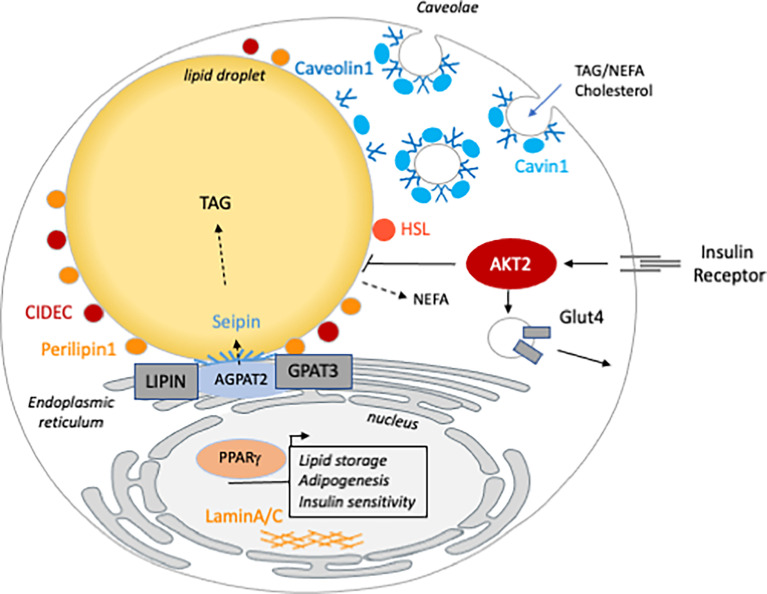
Lipodystrophies belong to the heterogenous group of syndromes in which the primary defect is a generalized or partial absence of adipose tissue, which may be congenital or acquired in origin. Lipodystrophy should be considered in patients manifesting the combination of insulin resistance (with or without overt diabetes), dyslipidemia and fatty liver. Lipodystrophies are classified according to the etiology of the disease (genetic or acquired) and to the anatomical distribution of adipose tissue (generalized or partial). The mechanism of adipose tissue loss is specific to each syndrome, depending on the biological function of the mutated gene. Mice models, together with cellular studies have permitted clarification of the mechanisms by which human mutations deeply compromise adipocyte homeostasis. In addition, rodent models have proven to be crucial in deciphering the cardiometabolic consequences of the lack of adipose tissue such as NAFLD, muscle insulin resistance and cardiomyopathy. More precisely, tissue-specific transgenic and knockout mice have brought new tools to distinguish phenotypic traits that are the consequences of lipodystrophy from those that are cell-autonomous. In this review, we discuss the mice models of lipodystrophy including those of inherited human syndromes of generalized and partial lipodystrophy. We present how these models have demonstrated the central role of white adipose tissue in energetic homeostasis in general, including insulin sensitivity and lipid handling in particular. We underscore the differences reported with the human phenotype and discuss the limit of rodent models in recapitulating adipose tissue primary default. Finally, we present how these mice models have highlighted the function of the causative-genes and brought new insights into the pathophysiology of the cardiometabolic complications associated with lipodystrophy.
脂肪营养不良属于一组异质性综合征,其主要缺陷是脂肪组织的普遍或部分缺失,可能是先天性的,也可能是后天获得的。当患者表现出胰岛素抵抗(伴有或不伴有显性糖尿病)、血脂异常和脂肪肝的组合时,应考虑脂肪营养不良。脂肪营养不良根据疾病的病因(遗传或后天获得)和脂肪组织的解剖分布(全身性或局部性)进行分类。脂肪组织丧失的机制因每个综合征而异,取决于突变基因的生物学功能。小鼠模型以及细胞研究使人们能够阐明人类基因突变如何严重破坏脂肪细胞的稳态的机制。此外,啮齿动物模型已被证明在阐明缺乏脂肪组织引起的代谢并发症(如非酒精性脂肪性肝病、肌肉胰岛素抵抗和心肌病)方面至关重要。更确切地说,组织特异性转基因和敲除小鼠为区分脂肪营养不良的表型特征与细胞自主性特征提供了新的工具。在这篇综述中,我们讨论了脂肪营养不良的小鼠模型,包括遗传性全身性和局部性脂肪营养不良的人类综合征。我们介绍了这些模型如何证明了白色脂肪组织在一般能量稳态中的核心作用,特别是在胰岛素敏感性和脂质处理方面。我们强调了与人类表型报告的差异,并讨论了啮齿动物模型在再现脂肪组织原发性缺陷方面的局限性。最后,我们介绍了这些小鼠模型如何突出了致病基因的功能,并为脂肪营养不良相关的心血管代谢并发症的病理生理学带来了新的见解。