Zhu Jiujun, Shen Yong, Wang Lina, Qiao Jianghua, Zhao Yajie, Wang Qiming
Department of Breast Disease, Henan Breast Cancer Center, Affiliated Cancer Hospital of Zhengzhou University, Henan Cancer Hospital, Zhengzhou, China.
Department of Clinical Laboratory, Affiliated Cancer Hospital of Zhengzhou University, Henan Cancer Hospital, Zhengzhou, China.
Ann Transl Med. 2022 Feb;10(3):143. doi: 10.21037/atm-21-6748.
The progression of breast cancer (BC) is highly dependent on the tumor microenvironment. Inflammation, stromal cells, and the immune landscape have been identified as significant drivers of BC in multiple preclinical studies. Therefore, this study aimed to clarify the predictive relevance of stromal and immune cell-associated genes in patients suffering from BC.
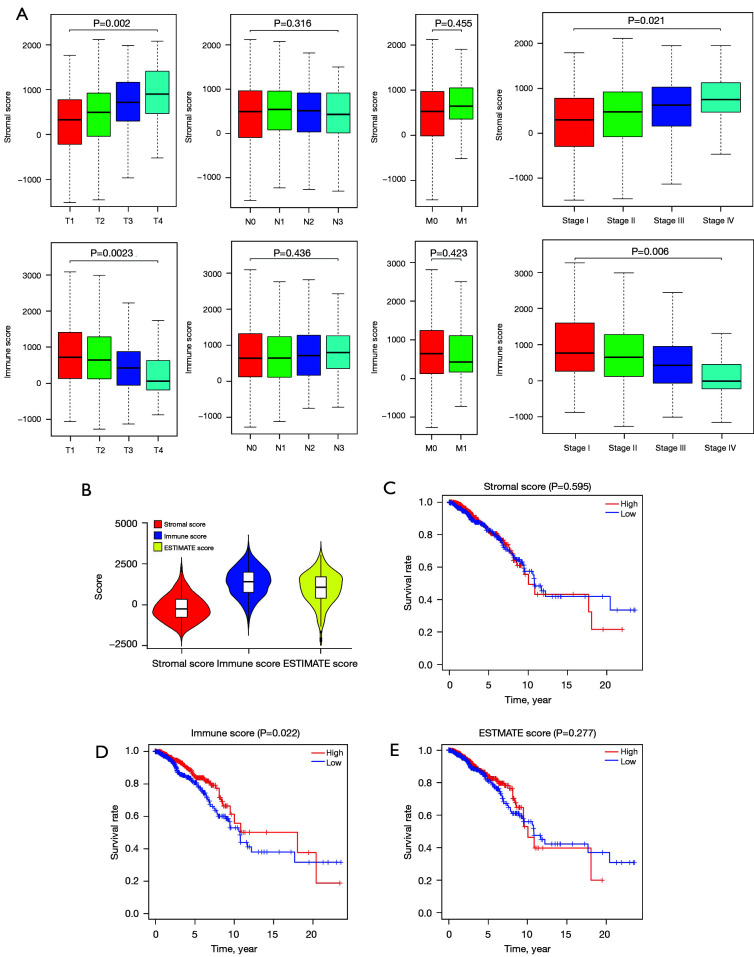
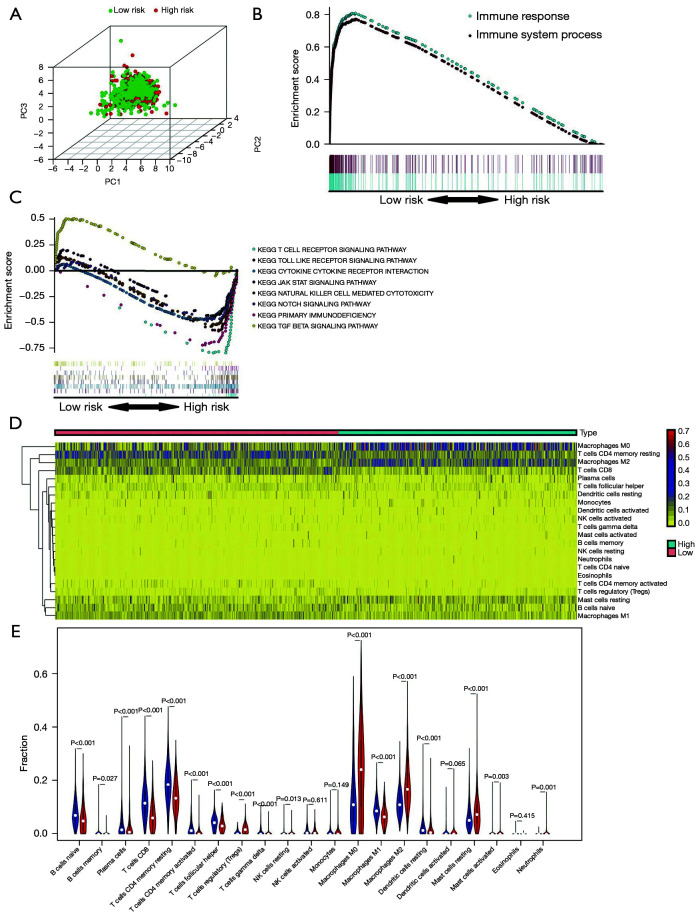
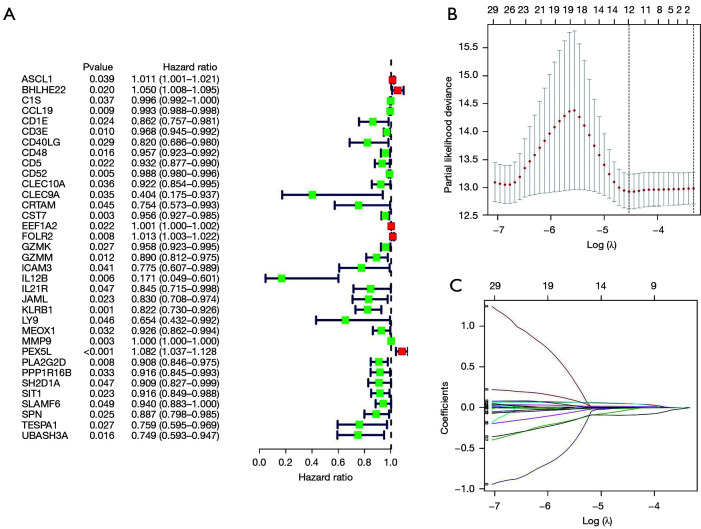
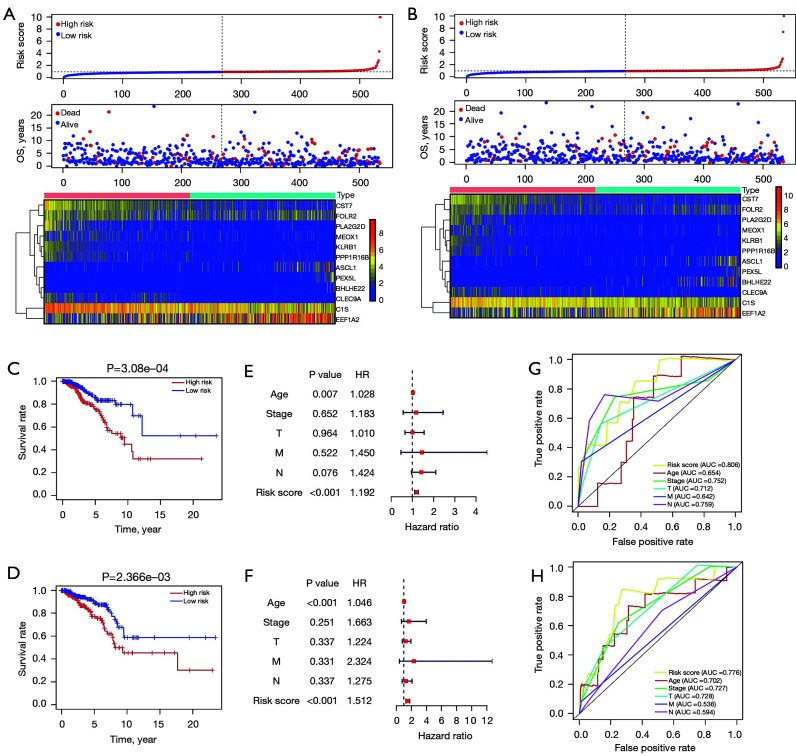
We employed the estimation of stromal and immune cells in malignant tumor tissues using expression data (ESTIMATE) algorithm to calculate the stromal and immunological scores, which were then used to evaluate differentially expressed genes (DEGs) in BC samples using The Cancer Genome Atlas (TCGA) database. Univariate analyses were conducted to identify the DEGs linked to survival in BC patients. Next, the prognostic DEGs (with a log-rank P<0.05) were used to create a risk signature, and the least absolute shrinkage and selection operator (LASSO) regression method was used to analyze and optimize the risk signature. The following formula was used to compute the prognostic risk score values: Risk score = Gene 1 * β1 + Gene 2 * β2 +… Gene n * βn. The median prognostic risk score values were used to divide BC patients into the low-risk (LR) and high-risk (HR) groups. The patient samples of the validation cohort were then assessed using this formula. We used principal component analysis (PCA) to determine the expression patterns of the different patient groups. Gene Set Enrichment Analysis (GSEA) was used to determine whether there were significant variations between the groups in the evaluated gene sets.
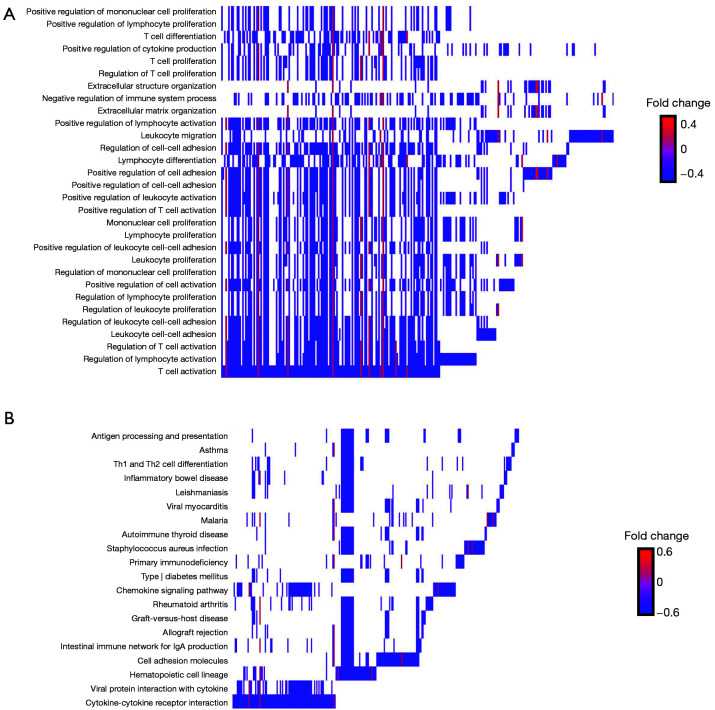
The present study revealed that DEGs linked with survival were closely associated with immunological responses. A prognostic signature was constructed that consisted of 12 genes ( and ). According to their survival, BC patients were separated into LR and HR groups using the identified 12-gene signature. The immunological status and immune cell infiltration were observed differently in the LR and HR groups.
Our results provide novel insights into several microenvironment-linked genes that influence survival outcomes in patients with BC, which suggests that these genes could be candidate therapeutic targets.
乳腺癌(BC)的进展高度依赖于肿瘤微环境。在多项临床前研究中,炎症、基质细胞和免疫格局已被确定为BC的重要驱动因素。因此,本研究旨在阐明基质和免疫细胞相关基因在BC患者中的预测相关性。
我们采用基于表达数据的恶性肿瘤组织中基质和免疫细胞估计(ESTIMATE)算法来计算基质和免疫评分,然后使用癌症基因组图谱(TCGA)数据库评估BC样本中的差异表达基因(DEG)。进行单变量分析以鉴定与BC患者生存相关的DEG。接下来,使用预后DEG(对数秩P<0.05)创建风险特征,并使用最小绝对收缩和选择算子(LASSO)回归方法分析和优化风险特征。使用以下公式计算预后风险评分值:风险评分=基因1β1+基因2β2+…基因n*βn。使用预后风险评分的中位数将BC患者分为低风险(LR)和高风险(HR)组。然后使用该公式评估验证队列的患者样本。我们使用主成分分析(PCA)来确定不同患者组的表达模式。基因集富集分析(GSEA)用于确定评估的基因集在组间是否存在显著差异。
本研究表明,与生存相关的DEG与免疫反应密切相关。构建了一个由12个基因组成的预后特征。根据生存情况,使用鉴定出的12基因特征将BC患者分为LR组和HR组。在LR组和HR组中观察到不同的免疫状态和免疫细胞浸润。
我们的结果为影响BC患者生存结果的几个与微环境相关的基因提供了新的见解,这表明这些基因可能是候选治疗靶点。