Department of General Chemistry, Belarusian State Medical University, Dzerzinskogo 83, Minsk, Belarus.
Biochemical Group of the Multidisciplinary Diagnostic Laboratory, Institute of Physiology of the National Academy of Sciences of Belarus, Minsk, Belarus.
Amino Acids. 2022 Aug;54(8):1155-1171. doi: 10.1007/s00726-022-03153-5. Epub 2022 Mar 16.
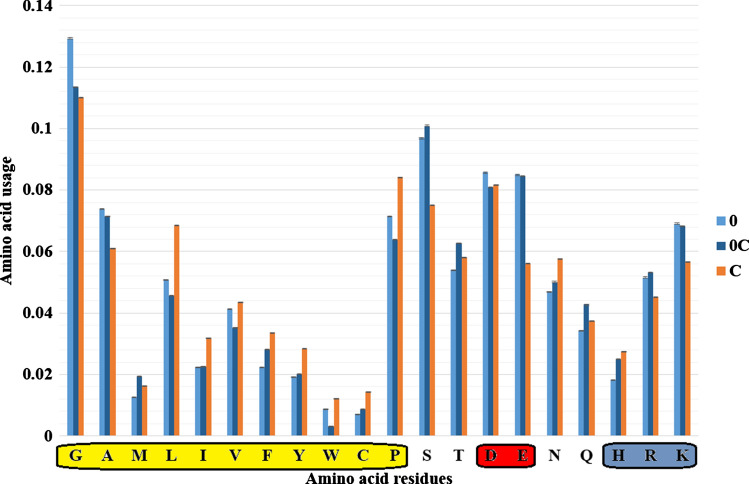
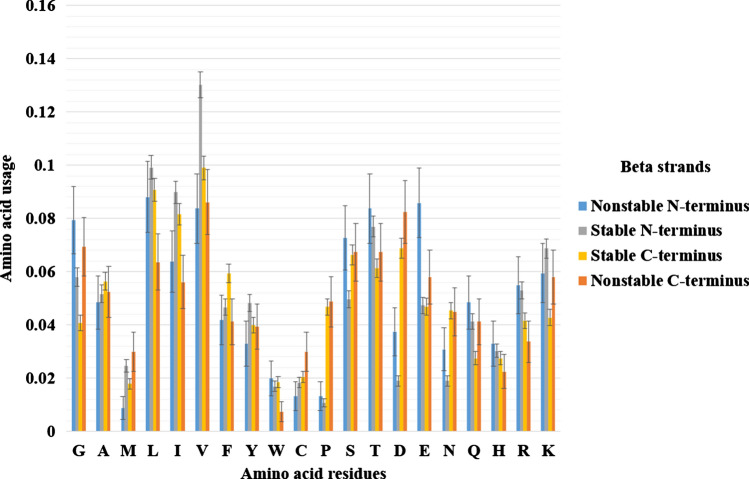
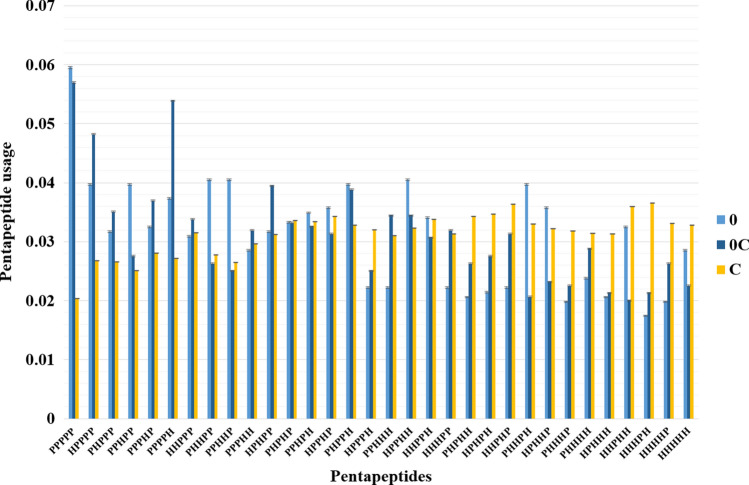
Intrinsically disordered proteins are frequently involved in important regulatory processes in the cell thanks to their ability to bind several different targets performing sometimes even opposite functions. The PentUnFOLD algorithm is a physicochemical method that is based on new propensity scales for disordered, nonstable and stable elements of secondary structure and on the counting of stabilizing and destabilizing intraprotein contacts. Unlike other methods, it works with a PDB file, and it can determine not only those fragments of alpha helices, beta strands, and random coils that can turn into disordered state (the "dark" side of the disorder), but also nonstable regions of alpha helices and beta strands which are able to turn into random coils (the "light" side), and vice versa (H ↔ C, E ↔ C). The scales have been obtained from structural data on disordered regions from the middle parts of amino acid sequences only, and not on their expectedly disordered N- and C-termini. Among other tendencies we have found that regions of both alpha helices and beta strands that can turn into the disordered state are relatively enriched in residues of Ala, Met, Asp, and Lys, while regions of both alpha helices and beta strands that can turn into random coil are relatively enriched in hydrophilic residues, and Cys, Pro, and Gly. Moreover, PentUnFOLD has the option to determine the effect of secondary structure transitions on the stability of a given region of a protein. The PentUnFOLD algorithm is freely available at http://3.17.12.213/pent-un-fold and http://chemres.bsmu.by/PentUnFOLD.htm .
无规卷曲蛋白质由于能够结合多个不同的靶标,执行有时甚至相反的功能,因此经常参与细胞中的重要调节过程。PentUnFOLD 算法是一种物理化学方法,它基于新的无规卷曲、非稳定和稳定二级结构元素的倾向性尺度,以及对稳定和不稳定的蛋白质内接触的计数。与其他方法不同,它使用 PDB 文件,可以确定不仅是那些可以变成无规状态的α螺旋、β链和无规卷曲片段(无规性的“暗”面),而且还可以确定能够变成无规卷曲的α螺旋和β链的非稳定区域(“亮”面),反之亦然(H ⇔ C,E ⇔ C)。这些尺度是从仅无序区域的氨基酸序列中间部分的结构数据中获得的,而不是从预期无序的 N-和 C-末端获得的。除了其他趋势,我们还发现可以变成无规状态的α螺旋和β链区域相对富含 Ala、Met、Asp 和 Lys 残基,而可以变成无规卷曲的α螺旋和β链区域相对富含亲水残基,Cys、Pro 和 Gly。此外,PentUnFOLD 还有一个选项可以确定二级结构转变对蛋白质特定区域稳定性的影响。PentUnFOLD 算法可在以下网址免费获取:http://3.17.12.213/pent-un-fold 和 http://chemres.bsmu.by/PentUnFOLD.htm 。