Alimohamed Mohamed Z, Westers Helga, Vos Yvonne J, Van der Velde K Joeri, Sijmons Rolf H, Van der Zwaag Paul A, Sikkema-Raddatz Birgit, Jongbloed Jan D H
Department of Genetics, University of Groningen, University Medical Center Groningen, Groningen, Netherlands.
Department of Haematology and Blood Transfusion, Muhimbili University of Health and Allied Sciences, Dar-es-Salaam, Tanzania.
Front Genet. 2022 Mar 1;13:824510. doi: 10.3389/fgene.2022.824510. eCollection 2022.
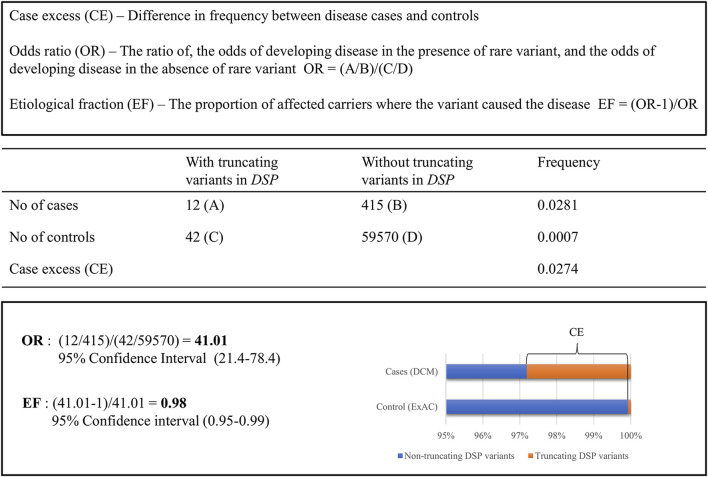
In the molecular genetic diagnostics of Mendelian disorders, solutions are needed for the major challenge of dealing with the large number of variants of uncertain significance (VUSs) identified using next-generation sequencing (NGS). Recently, promising approaches using constraint metrics to calculate case excess scores (CE), etiological fractions (EF), and gnomAD-derived constraint scores have been reported that estimate the likelihood of rare variants in specific genes or regions that are pathogenic. Our objective is to study the usability of these constraint data into variant interpretation in a diagnostic setting, using our cardiomyopathy cohort. Patients (N = 2002) referred for clinical genetic diagnostics underwent NGS testing of 55-61 genes associated with cardiomyopathies. Previously classified likely pathogenic (LP) and pathogenic (P) variants were used to validate the use of data from CE, EF, and gnomAD constraint analyses for (re)classification of associated variant types in specific cardiomyopathy subtype-related genes. The classifications corroborated in 94% (354/378) of cases. Next, we reclassified 23 unique VUSs to LP, increasing the diagnostic yield by 1.2%. In addition, 106 unique VUSs (5.3% of patients) were prioritized for co-segregation or functional analyses. Our analysis confirms that the use of constraint metrics data can improve variant interpretation, and we, therefore, recommend using constraint scores on other cohorts and disorders and its inclusion in variant interpretation protocols.
在孟德尔疾病的分子遗传学诊断中,需要解决使用下一代测序(NGS)识别出的大量意义未明变异(VUS)这一重大挑战的解决方案。最近,有报道称使用约束指标来计算病例超额分数(CE)、病因分数(EF)和gnomAD衍生的约束分数的有前景的方法,这些方法可估计特定基因或区域中罕见变异的致病可能性。我们的目标是使用我们的心肌病队列研究这些约束数据在诊断环境中用于变异解读的实用性。因临床基因诊断而转诊的患者(N = 2002)接受了与心肌病相关的55 - 61个基因的NGS检测。先前分类为可能致病(LP)和致病(P)的变异用于验证CE、EF和gnomAD约束分析数据在特定心肌病亚型相关基因中相关变异类型(重新)分类的用途。在94%(354/378)的病例中得到了证实。接下来,我们将23个独特的VUS重新分类为LP,诊断率提高了1.2%。此外,对106个独特的VUS(占患者的5.3%)进行了共分离或功能分析的优先级排序。我们的分析证实,使用约束指标数据可以改善变异解读,因此,我们建议在其他队列和疾病中使用约束分数,并将其纳入变异解读方案。