Center for Algorithmic Biotechnology, Institute of Translational Biomedicine, St. Petersburg State University, St. Petersburg 199034, Russia.
Brain and Mind Research Institute and Center for Neurogenetics, Weill Cornell Medicine, New York, New York 10065, USA.
Genome Res. 2022 Apr;32(4):726-737. doi: 10.1101/gr.276405.121. Epub 2022 Mar 17.
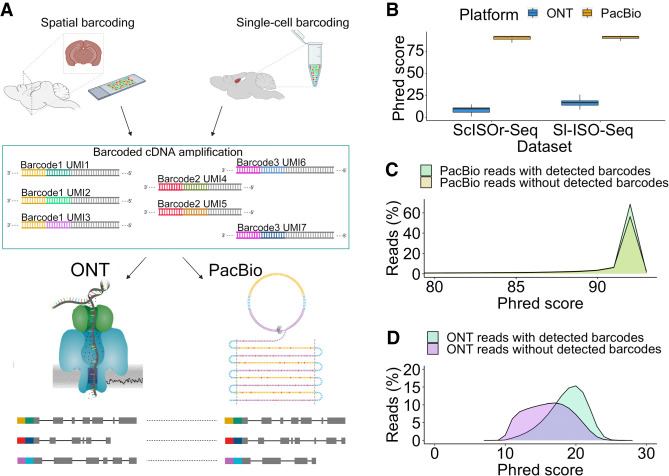
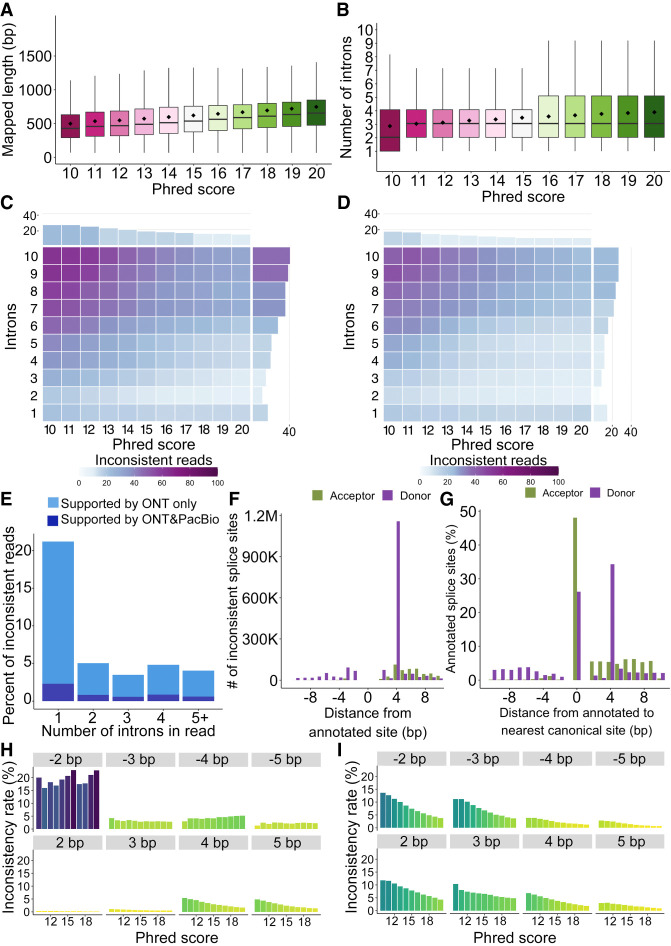
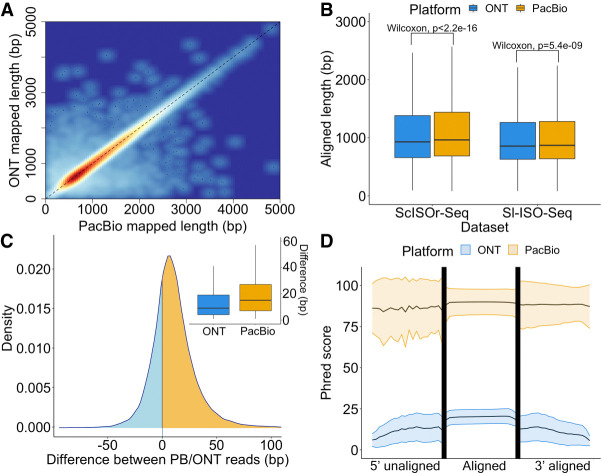
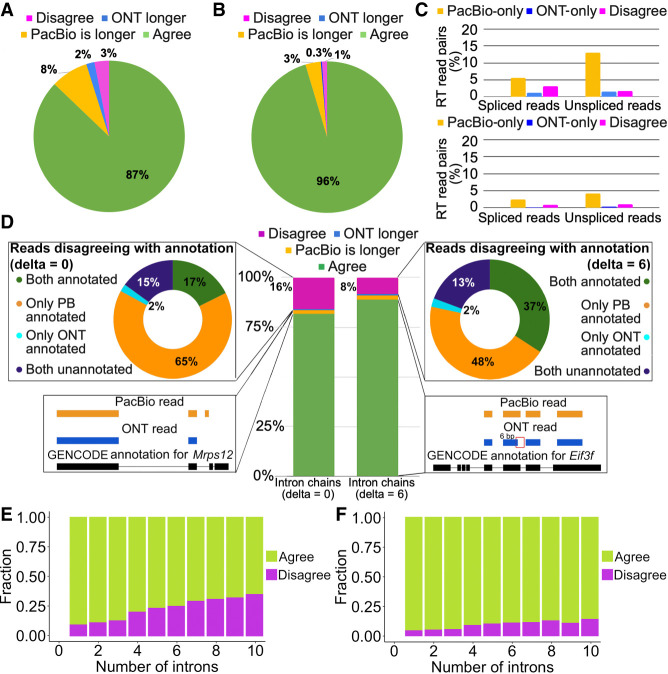
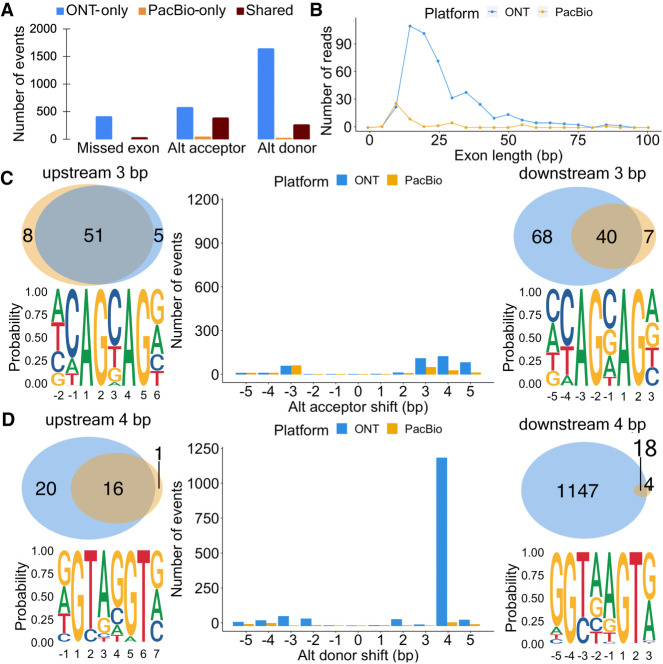
Long-read transcriptomics require understanding error sources inherent to technologies. Current approaches cannot compare methods for an individual RNA molecule. Here, we present a novel platform-comparison method that combines barcoding strategies and long-read sequencing to sequence cDNA copies representing an individual RNA molecule on both Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT). We compare these long-read pairs in terms of sequence content and isoform patterns. Although individual read pairs show high similarity, we find differences in (1) aligned length, (2) transcription start site (TSS), (3) polyadenylation site (poly(A)-site) assignment, and (4) exon-intron structures. Overall, 25% of read pairs disagree on either TSS, poly(A)-site, or splice site. Intron-chain disagreement typically arises from alignment errors of microexons and complicated splice sites. Our single-molecule technology comparison reveals that inconsistencies are often caused by sequencing error-induced inaccurate ONT alignments, especially to downstream GUNNGU donor motifs. However, annotation-disagreeing upstream shifts in NAGNAG acceptors in ONT are often confirmed by PacBio and are thus likely real. In both barcoded and nonbarcoded ONT reads, we find that intron number and proximity of GU/AGs better predict inconsistencies with the annotation than read quality alone. We summarize these findings in an annotation-based algorithm for spliced alignment correction that improves subsequent transcript construction with ONT reads.
长读转录组学需要了解技术固有的误差源。目前的方法无法比较单个 RNA 分子的方法。在这里,我们提出了一种新的平台比较方法,该方法结合了条形码策略和长读测序,以对单个 RNA 分子的 cDNA 拷贝进行测序,这些拷贝代表了太平洋生物科学公司(PacBio)和牛津纳米孔技术公司(ONT)的两种技术。我们根据序列内容和同工型模式对这些长读对进行比较。尽管单个读对显示出高度的相似性,但我们发现了以下差异:(1)比对长度,(2)转录起始位点(TSS),(3)多聚腺苷酸化位点(poly(A)-site)分配,以及(4)外显子-内含子结构。总体而言,25%的读对在 TSS、poly(A)-site 或剪接位点上存在分歧。内含子链的分歧通常是由于微外显子和复杂剪接位点的比对错误引起的。我们的单分子技术比较表明,不一致通常是由测序错误引起的 ONT 不准确比对引起的,尤其是下游 GUNNGU 供体基序。然而,ONT 中注释不一致的上游 NAGNAG 受体的移位通常被 PacBio 证实,因此可能是真实的。在带条形码和不带条形码的 ONT 读取中,我们发现,内含子数量和 GU/AG 的接近程度比单独的读取质量更能预测与注释的不一致性。我们将这些发现总结在一个基于注释的剪接对齐校正算法中,该算法可以提高使用 ONT 读取进行后续转录本构建的准确性。