Siolta Therapeutics, 930 Brittan Ave, San Carlos, CA 94070, USA.
Pacific Biosciences, 1305 O'Brien Dr, Menlo Park, CA 93025, USA.
Microb Genom. 2022 Mar;8(3). doi: 10.1099/mgen.0.000794.
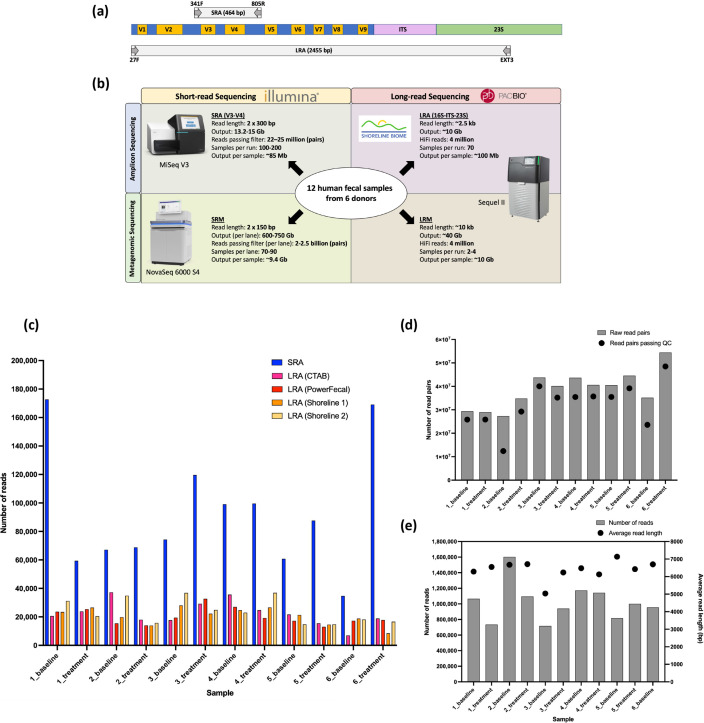
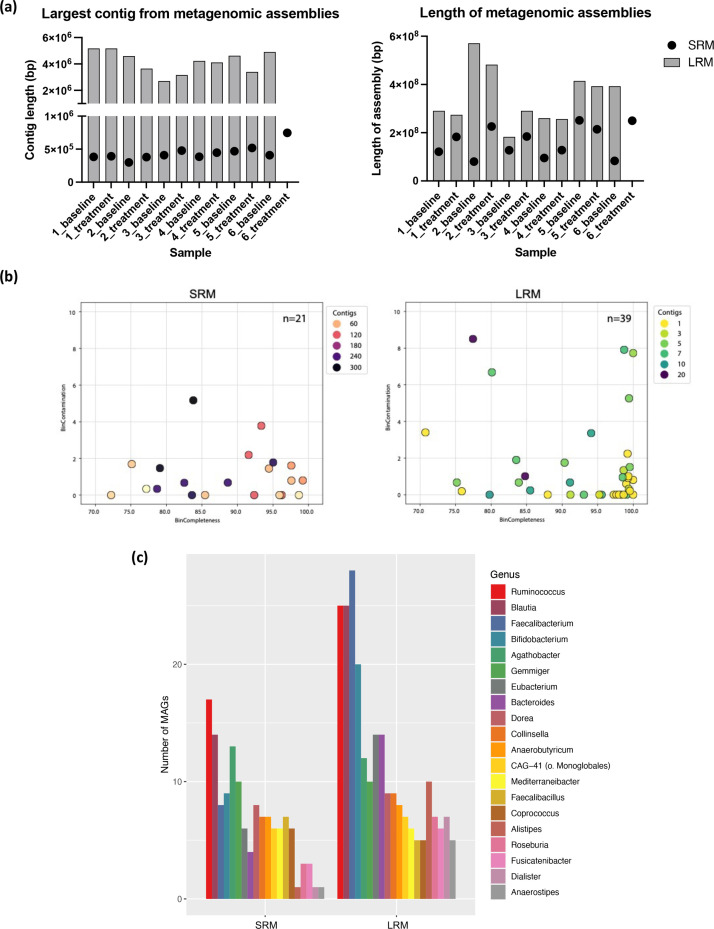
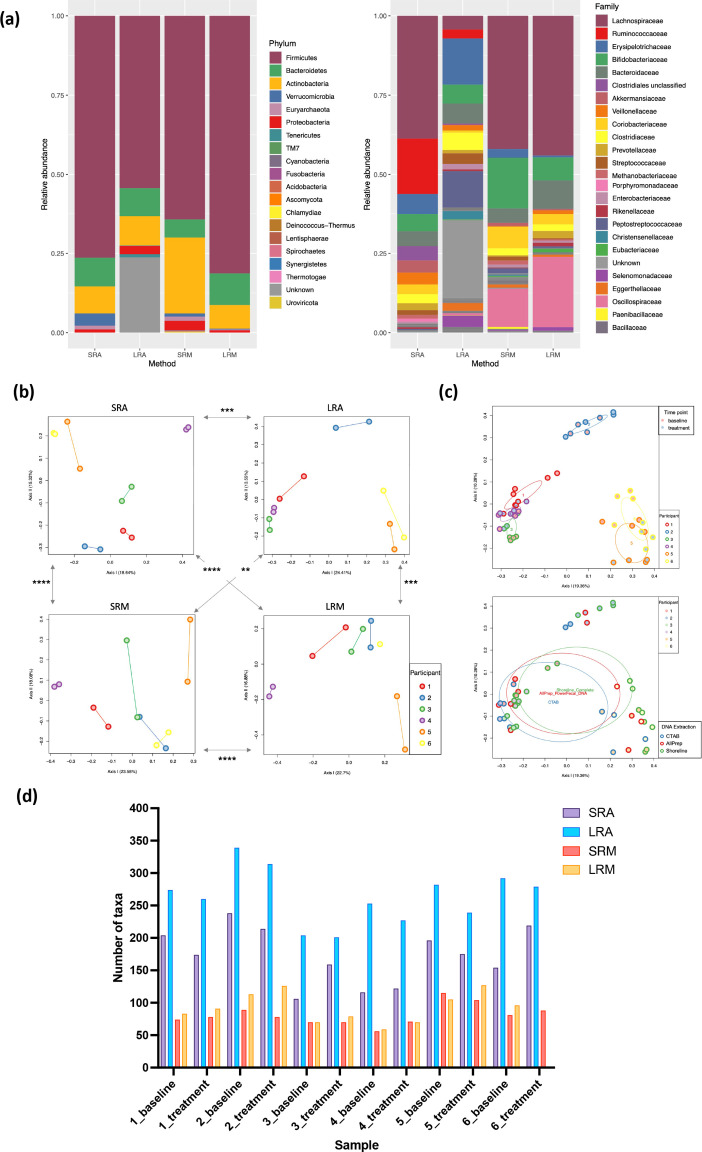
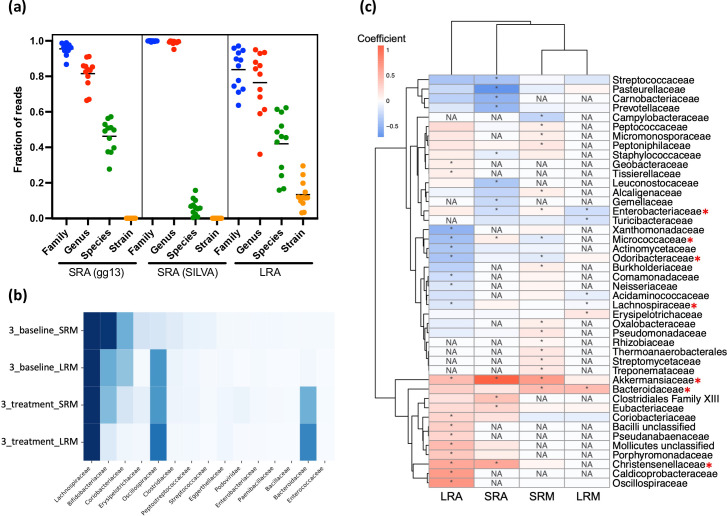
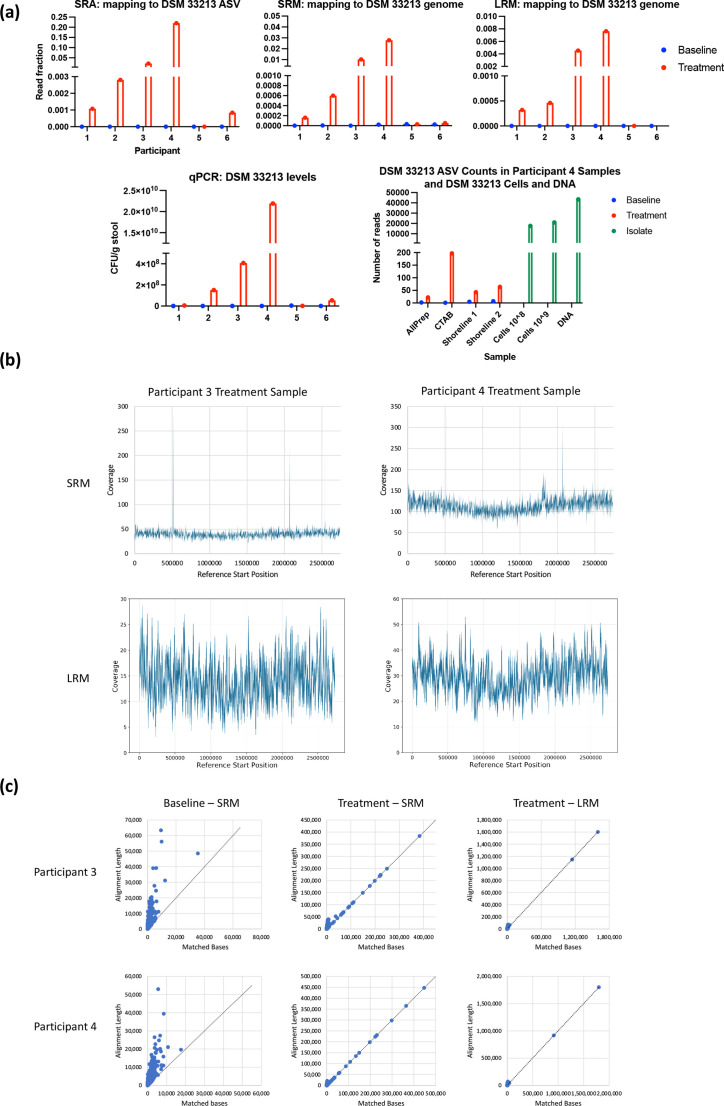
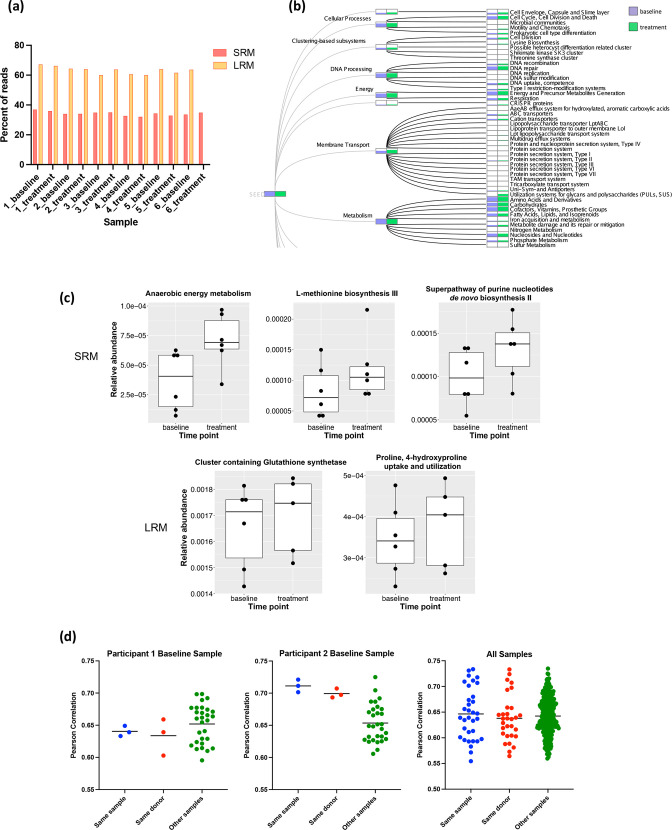
A long-standing challenge in human microbiome research is achieving the taxonomic and functional resolution needed to generate testable hypotheses about the gut microbiota's impact on health and disease. With a growing number of live microbial interventions in clinical development, this challenge is renewed by a need to understand the pharmacokinetics and pharmacodynamics of therapeutic candidates. While short-read sequencing of the bacterial 16S rRNA gene has been the standard for microbiota profiling, recent improvements in the fidelity of long-read sequencing underscores the need for a re-evaluation of the value of distinct microbiome-sequencing approaches. We leveraged samples from participants enrolled in a phase 1b clinical trial of a novel live biotherapeutic product to perform a comparative analysis of short-read and long-read amplicon and metagenomic sequencing approaches to assess their utility for generating clinical microbiome data. Across all methods, overall community taxonomic profiles were comparable and relationships between samples were conserved. Comparison of ubiquitous short-read 16S rRNA amplicon profiling to long-read profiling of the 16S-ITS-23S rRNA amplicon showed that only the latter provided strain-level community resolution and insight into novel taxa. All methods identified an active ingredient strain in treated study participants, though detection confidence was higher for long-read methods. Read coverage from both metagenomic methods provided evidence of active-ingredient strain replication in some treated participants. Compared to short-read metagenomics, approximately twice the proportion of long reads were assigned functional annotations. Finally, compositionally similar bacterial metagenome-assembled genomes (MAGs) were recovered from short-read and long-read metagenomic methods, although a greater number and more complete MAGs were recovered from long reads. Despite higher costs, both amplicon and metagenomic long-read approaches yielded added microbiome data value in the form of higher confidence taxonomic and functional resolution and improved recovery of microbial genomes compared to traditional short-read methodologies.
人类微生物组研究中长期存在的挑战是实现分类和功能分辨率,以生成关于肠道微生物组对健康和疾病影响的可测试假设。随着越来越多的活体微生物干预措施在临床开发中,这一挑战因需要了解治疗候选物的药代动力学和药效学而重新出现。虽然细菌 16S rRNA 基因的短读测序一直是微生物组分析的标准,但长读测序保真度的提高突显了重新评估不同微生物组测序方法价值的必要性。我们利用参加新型活体生物治疗产品 1b 期临床试验的参与者的样本,对短读和长读扩增子和宏基因组测序方法进行了比较分析,以评估它们在生成临床微生物组数据方面的实用性。在所有方法中,总体群落分类特征相似,样本之间的关系得以保留。普遍存在的短读 16S rRNA 扩增子分析与 16S-ITS-23S rRNA 扩增子的长读分析比较表明,只有后者提供了种群水平的社区分辨率,并深入了解了新的分类群。所有方法都在接受治疗的研究参与者中鉴定出了活性成分菌株,但长读方法的检测置信度更高。两种宏基因组方法的读取覆盖率都提供了一些治疗参与者中活性成分菌株复制的证据。与短读宏基因组学相比,长读方法分配功能注释的比例约为两倍。最后,从短读和长读宏基因组方法中回收了组成相似的细菌宏基因组组装基因组(MAGs),尽管长读方法回收的 MAGs 数量更多且更完整。尽管成本较高,但与传统的短读方法相比,扩增子和宏基因组长读方法在提高分类和功能分辨率方面提供了更多的微生物组数据价值,并提高了微生物基因组的恢复率。