Shibuya Yoshihiro, Belazzougui Djamal, Kucherov Gregory
LIGM, Université Gustave Eiffel, Marne-la-Vallée, France.
CAPA, DTISI, Centre de Recherche sur l'Information Scientifique et Technique, Algiers, DZ, Algeria.
Algorithms Mol Biol. 2022 Mar 21;17(1):5. doi: 10.1186/s13015-022-00212-0.
k-mer counting is a common task in bioinformatic pipelines, with many dedicated tools available. Many of these tools produce in output k-mer count tables containing both k-mers and counts, easily reaching tens of GB. Furthermore, such tables do not support efficient random-access queries in general.
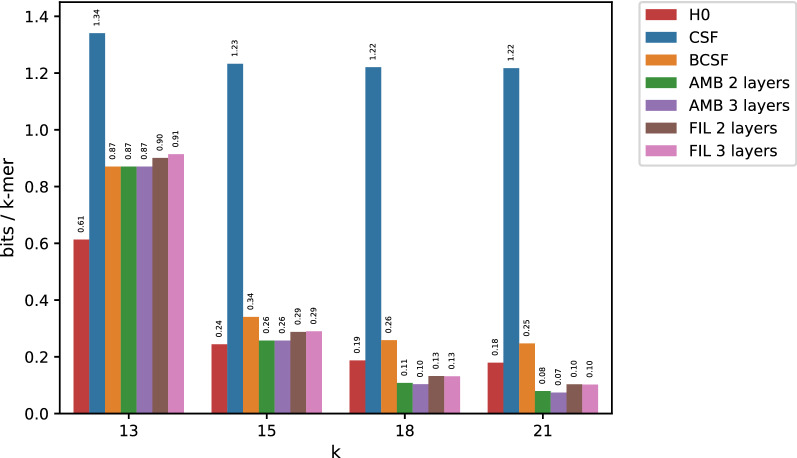
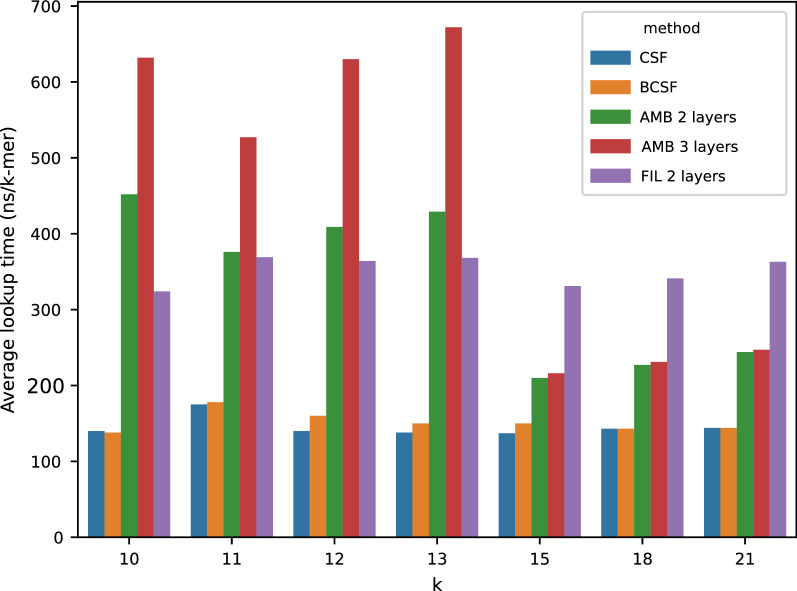
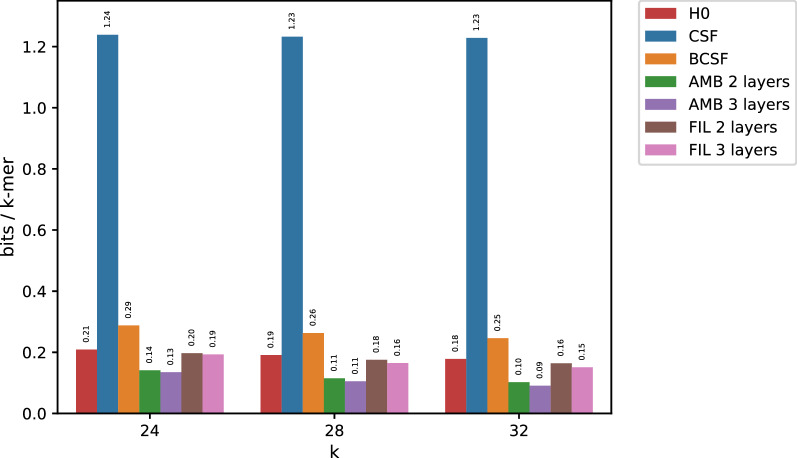
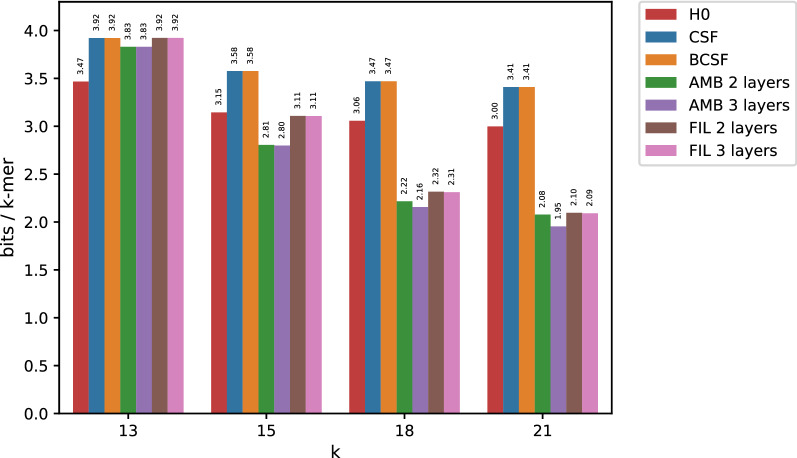
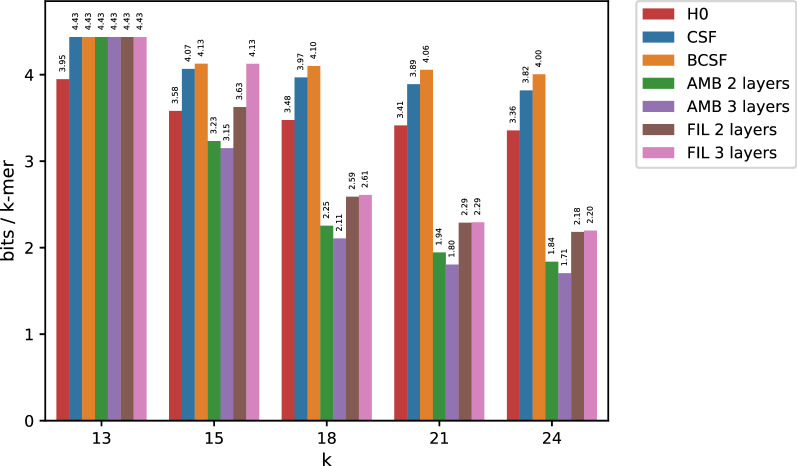
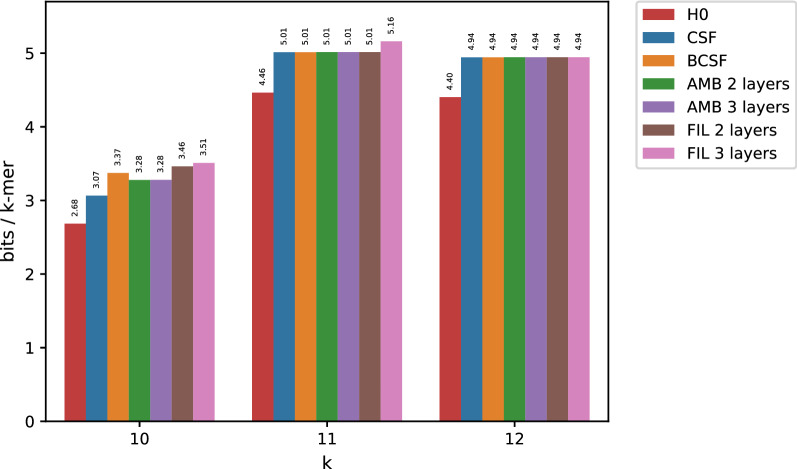
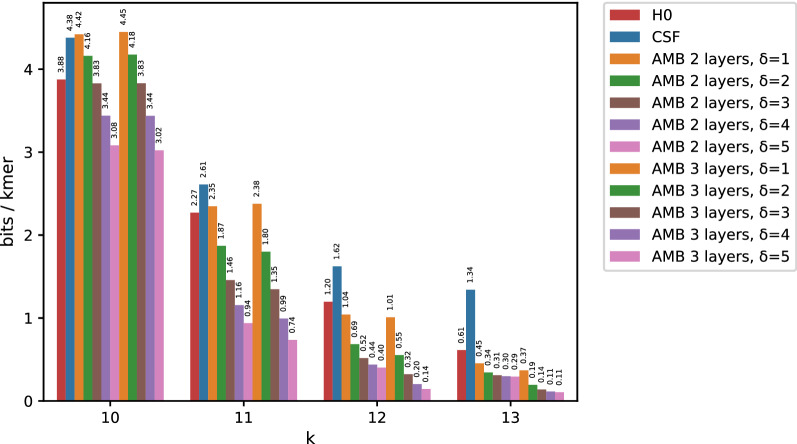
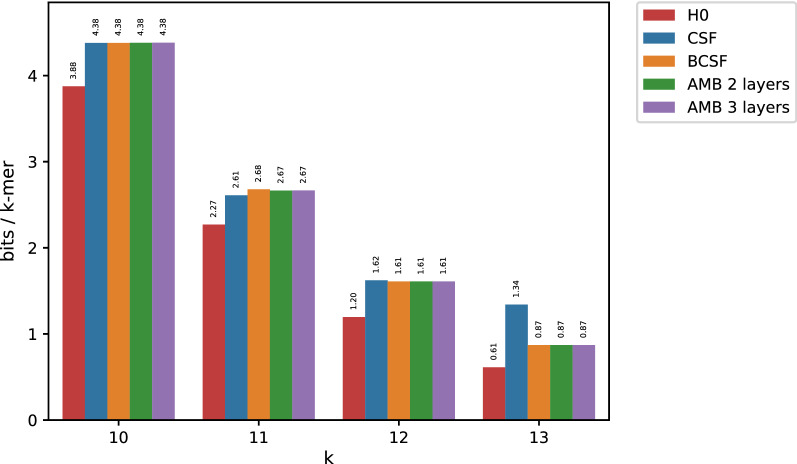
In this work, we design an efficient representation of k-mer count tables supporting fast random-access queries. We propose to apply Compressed Static Functions (CSFs), with space proportional to the empirical zero-order entropy of the counts. For very skewed distributions, like those of k-mer counts in whole genomes, the only currently available implementation of CSFs does not provide a compact enough representation. By adding a Bloom filter to a CSF we obtain a Bloom-enhanced CSF (BCSF) effectively overcoming this limitation. Furthermore, by combining BCSFs with minimizer-based bucketing of k-mers, we build even smaller representations breaking the empirical entropy lower bound, for large enough k. We also extend these representations to the approximate case, gaining additional space. We experimentally validate these techniques on k-mer count tables of whole genomes (E. Coli and C. Elegans) and unassembled reads, as well as on k-mer document frequency tables for 29 E. Coli genomes. In the case of exact counts, our representation takes about a half of the space of the empirical entropy, for large enough k's.
k-mer计数是生物信息学流程中的一项常见任务,有许多专用工具可用。这些工具中的许多都会生成包含k-mer和计数的输出k-mer计数值表,其大小很容易达到数十GB。此外,一般来说,这样的表不支持高效的随机访问查询。
在这项工作中,我们设计了一种k-mer计数值表的高效表示方法,支持快速随机访问查询。我们建议应用压缩静态函数(CSF),其空间与计数的经验零阶熵成正比。对于非常偏斜的分布,比如全基因组中的k-mer计数分布,目前唯一可用的CSF实现并不能提供足够紧凑的表示。通过在CSF中添加布隆过滤器,我们得到了布隆增强CSF(BCSF),有效地克服了这一限制。此外,通过将BCSF与基于最小化器的k-mer分桶相结合,对于足够大的k,我们构建了更小的表示,打破了经验熵下限。我们还将这些表示扩展到近似情况,从而获得更多空间。我们在全基因组(大肠杆菌和秀丽隐杆线虫)和未组装读数的k-mer计数值表以及29个大肠杆菌基因组的k-mer文档频率表上对这些技术进行了实验验证。在精确计数的情况下,对于足够大的k,我们的表示占用的空间约为经验熵的一半。