Department of Pharmacology and Cancer Biology and Duke Cancer Institute, Duke University, Durham, NC 27710, USA.
Department of Pathology, Duke University, Durham, NC 27710, USA.
Sci Transl Med. 2022 Mar 30;14(638):eabc7480. doi: 10.1126/scitranslmed.abc7480.
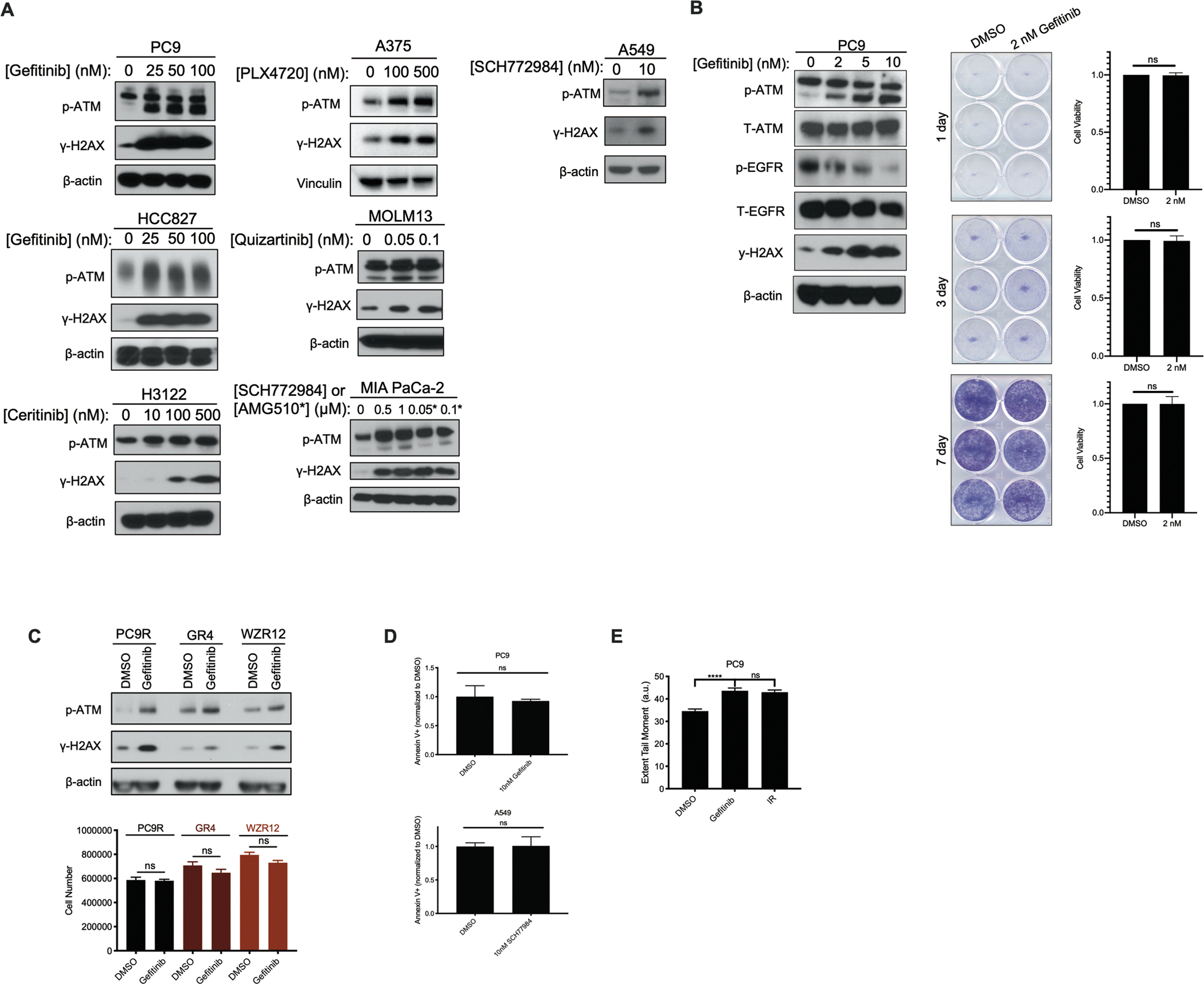
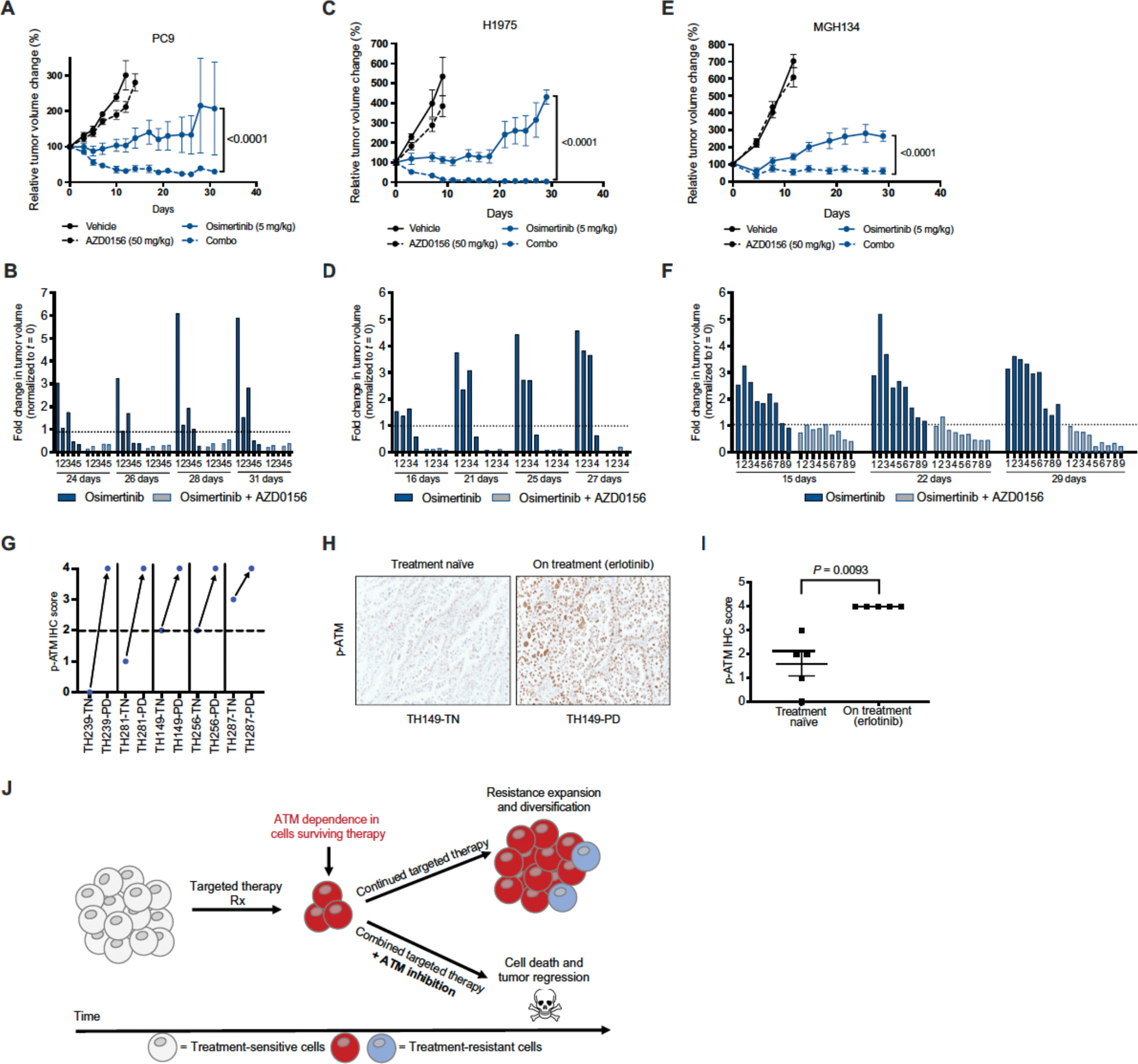
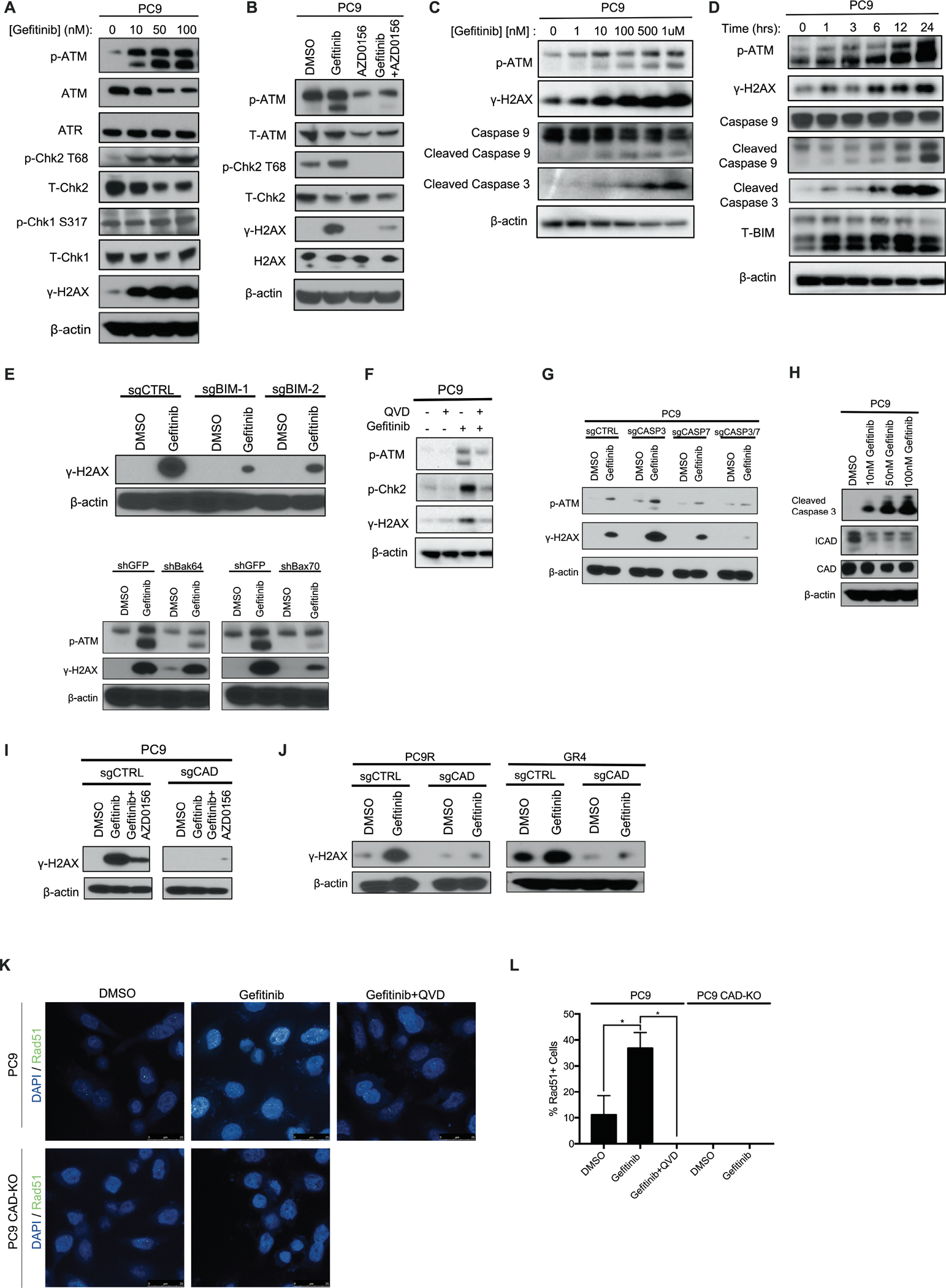
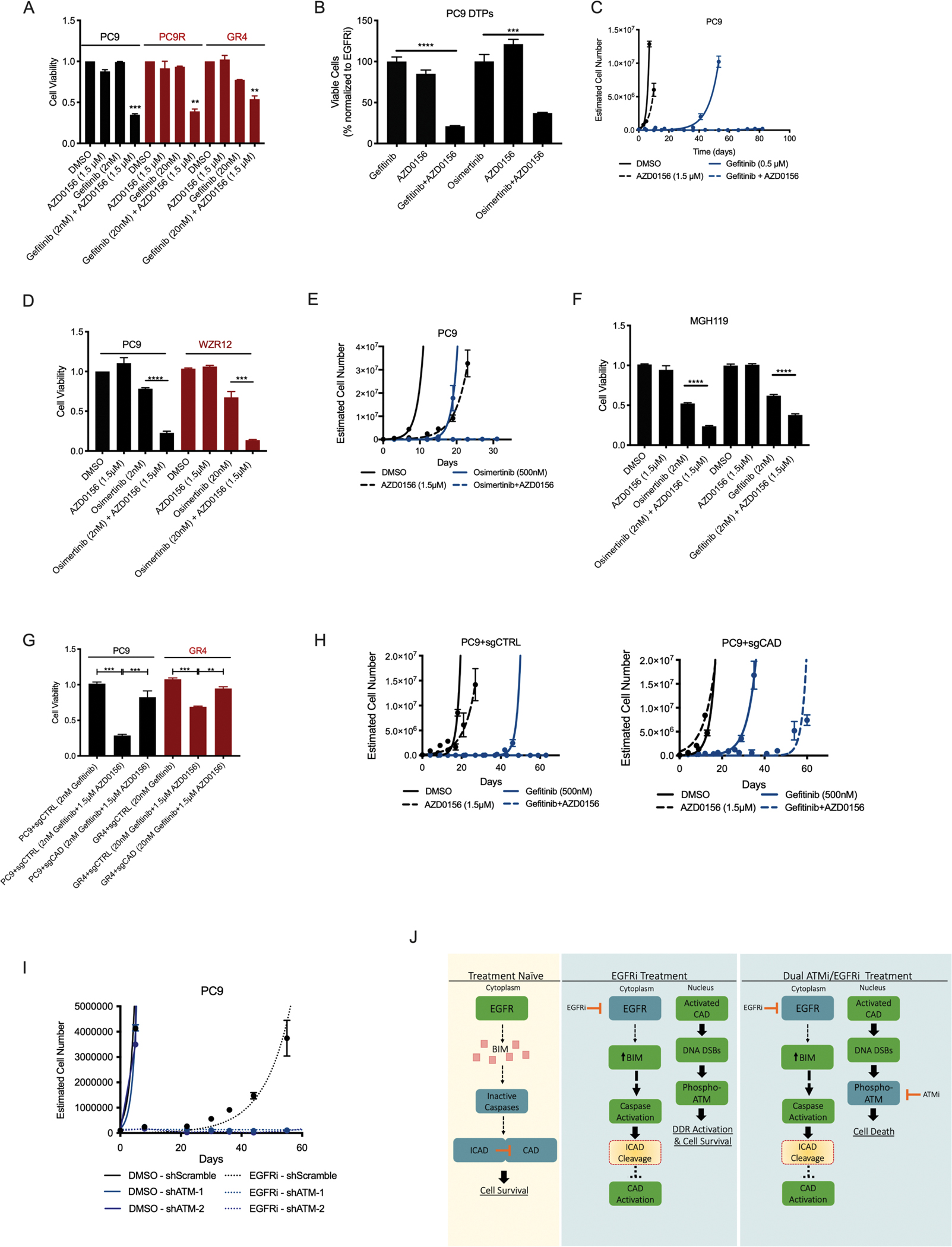
Residual cancer cells that survive drug treatments with targeted therapies act as a reservoir from which eventual resistant disease emerges. Although there is great interest in therapeutically targeting residual cells, efforts are hampered by our limited knowledge of the vulnerabilities existing in this cell state. Here, we report that diverse oncogene-targeted therapies, including inhibitors of epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), KRAS, and BRAF, induce DNA double-strand breaks and, consequently, ataxia-telangiectasia mutated (ATM)-dependent DNA repair in oncogene-matched residual tumor cells. This DNA damage response, observed in cell lines, mouse xenograft models, and human patients, is driven by a pathway involving the activation of caspases 3 and 7 and the downstream caspase-activated deoxyribonuclease (CAD). CAD is, in turn, activated through caspase-mediated degradation of its endogenous inhibitor, ICAD. In models of mutant non-small cell lung cancer (NSCLC), tumor cells that survive treatment with small-molecule EGFR-targeted therapies are thus synthetically dependent on ATM, and combined treatment with an ATM kinase inhibitor eradicates these cells in vivo. This led to more penetrant and durable responses in mutant NSCLC mouse xenograft models, including those derived from both established cell lines and patient tumors. Last, we found that rare patients with mutant NSCLC harboring co-occurring, loss-of-function mutations in exhibit extended progression-free survival on first generation EGFR inhibitor therapy relative to patients with mutant NSCLC lacking deleterious mutations. Together, these findings establish a rationale for the mechanism-based integration of ATM inhibitors alongside existing targeted therapies.
残留的癌细胞在接受靶向治疗药物治疗后存活下来,成为耐药疾病最终出现的源泉。尽管人们对靶向治疗残留细胞有很大的兴趣,但由于我们对这种细胞状态下存在的脆弱性了解有限,因此努力受到了阻碍。在这里,我们报告说,各种致癌基因靶向治疗,包括表皮生长因子受体(EGFR)、间变性淋巴瘤激酶(ALK)、KRAS 和 BRAF 抑制剂,会诱导 DNA 双链断裂,并因此导致与致癌基因匹配的残留肿瘤细胞中共济失调毛细血管扩张突变(ATM)依赖性 DNA 修复。这种在细胞系、小鼠异种移植模型和人类患者中观察到的 DNA 损伤反应,是由涉及半胱天冬酶 3 和 7 的激活以及下游半胱天冬酶激活的脱氧核糖核酸酶(CAD)的途径驱动的。CAD 反过来又通过半胱天冬酶介导的其内源性抑制剂 ICAD 的降解而被激活。在突变型非小细胞肺癌(NSCLC)模型中,经过小分子 EGFR 靶向治疗存活下来的肿瘤细胞因此在合成上依赖于 ATM,并且联合使用 ATM 激酶抑制剂可以在体内根除这些细胞。这导致突变型 NSCLC 小鼠异种移植模型中更具渗透性和更持久的反应,包括来自已建立的细胞系和患者肿瘤的模型。最后,我们发现少数携带同时发生的、功能丧失突变的突变型 NSCLC 患者在接受第一代 EGFR 抑制剂治疗时表现出无进展生存期延长,而缺乏有害突变的突变型 NSCLC 患者则没有。总之,这些发现为基于机制的 ATM 抑制剂与现有靶向治疗的整合提供了依据。