Department of Stomatology, Beijing Chaoyang Hospital, Capital Medical University, 8 Gongti Nan Lu, Chaoyang District, Beijing, 100020, China.
Department of Periodontology, Tianjin Stomatological Hospital, School of Medicine, Nankai University, Tianjin, China.
BMC Oral Health. 2022 Apr 9;22(1):118. doi: 10.1186/s12903-022-02150-0.
Periodontitis is a complex infectious disease with various causes and contributing factors. The aim of this study was to identify key genes, microRNAs (miRNAs) and transcription factors (TFs) and construct a miRNA-mRNA-TF regulatory networks to investigate the underlying molecular mechanism in periodontitis.
The GSE54710 miRNA microarray dataset and the gene expression microarray dataset GSE16134 were downloaded from the Gene Expression Omnibus database. The differentially expressed miRNAs (DEMis) and mRNAs (DEMs) were screened using the "limma" package in R. The intersection of the target genes of candidate DEMis and DEMs were considered significant DEMs in the regulatory network. Next, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted. Subsequently, DEMs were uploaded to the STRING database, a protein-protein interaction (PPI) network was established, and the cytoHubba and MCODE plugins were used to screen out key hub mRNAs and significant modules. Ultimately, to investigate the regulatory network underlying periodontitis, a global triple network including miRNAs, mRNAs, and TFs was constructed using Cytoscape software.
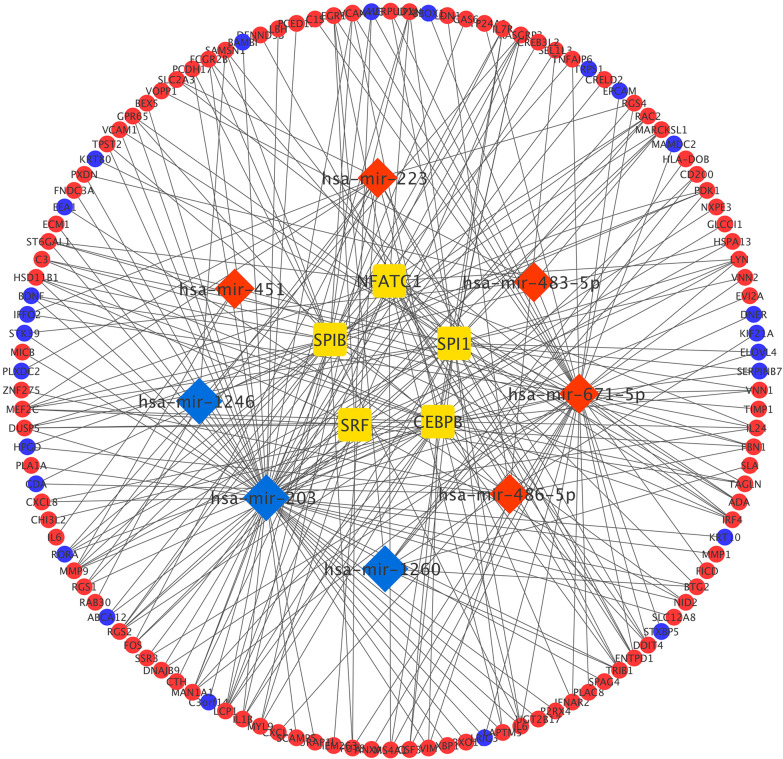
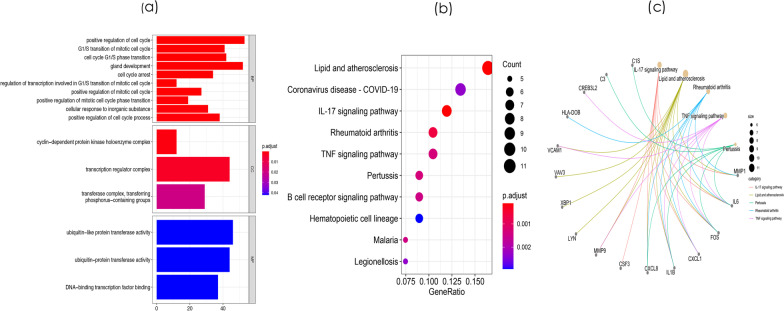
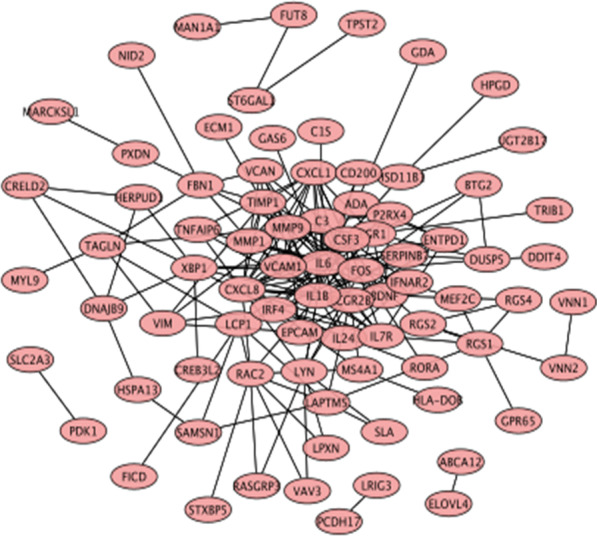
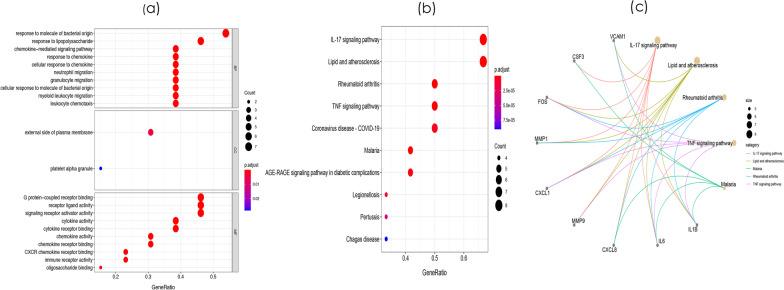
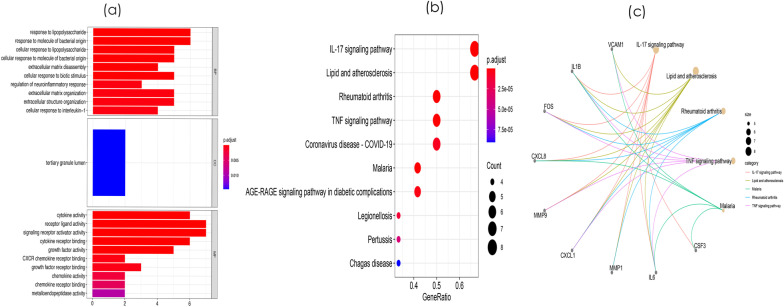
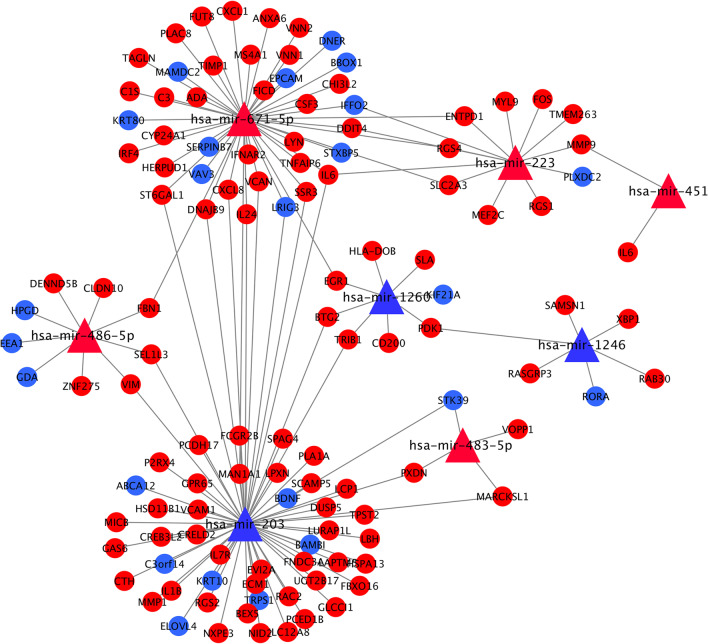
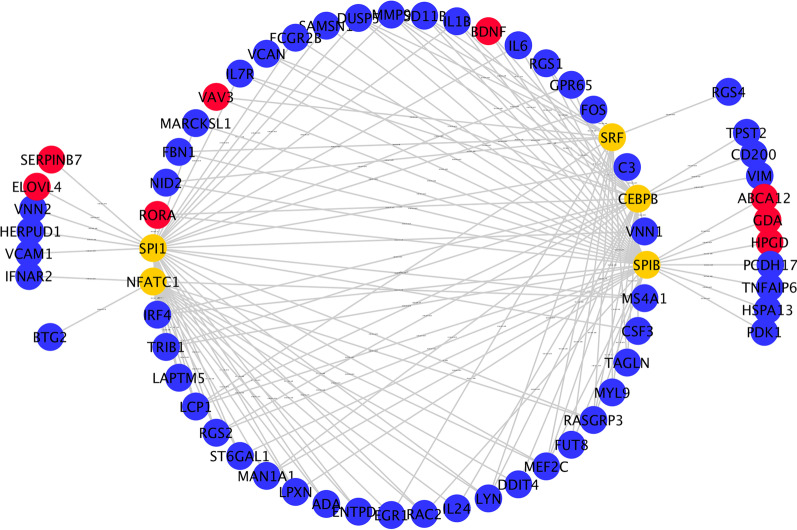
8 DEMis and 121 DEMs were found between the periodontal and control groups. GO analysis showed that mRNAs were most significantly enriched in positive regulation of the cell cycle, and KEGG pathway analysis showed that mRNAs in the regulatory network were mainly involved in the IL-17 signalling pathway. A PPI network was constructed including 81 nodes and 414 edges. Furthermore, 12 hub genes ranked by the top 10% genes with high degree connectivity and five TFs, including SRF, CNOT4, SIX6, SRRM3, NELFA, and ONECUT3, were identified and might play crucial roles in the molecular pathogenesis of periodontitis. Additionally, a miRNA-mRNA-TF coregulatory network was established.
In this study, we performed an integrated analysis based on public databases to identify specific TFs, miRNAs, and mRNAs that may play a pivotal role in periodontitis. On this basis, a TF-miRNA-mRNA network was established to provide a comprehensive perspective of the regulatory mechanism networks of periodontitis.
牙周炎是一种病因复杂、多因素参与的感染性疾病。本研究旨在识别关键基因、微小 RNA(miRNA)和转录因子(TF),构建 miRNA-mRNA-TF 调控网络,探讨牙周炎的潜在分子机制。
从基因表达综合数据库(GEO)中下载 GSE54710 miRNA 芯片数据集和 GSE16134 基因表达微阵列数据集。使用 R 中的“limma”包筛选差异表达的 miRNAs(DEMis)和 mRNAs(DEMs)。候选 DEMis 和 DEMs 的靶基因的交集被认为是调控网络中的显著 DEMs。接下来,进行基因本体论(GO)和京都基因与基因组百科全书(KEGG)通路富集分析。随后,将 DEMs 上传至 STRING 数据库,构建蛋白质-蛋白质相互作用(PPI)网络,使用 cytoHubba 和 MCODE 插件筛选关键 hub mRNAs 和显著模块。最终,使用 Cytoscape 软件构建包括 miRNAs、mRNAs 和 TFs 的全局三重网络,构建牙周炎调控网络。
牙周炎组和对照组之间发现了 8 个 DEMis 和 121 个 DEMs。GO 分析表明,mRNAs 最显著富集于细胞周期的正调控,KEGG 通路分析表明,调控网络中的 mRNAs 主要参与 IL-17 信号通路。构建了一个包括 81 个节点和 414 个边的 PPI 网络。此外,通过高连接度的前 10%基因排名确定了 12 个枢纽基因和 5 个 TF,包括 SRF、CNOT4、SIX6、SRRM3、NELFA 和 ONECUT3,它们可能在牙周炎的分子发病机制中发挥关键作用。此外,建立了 miRNA-mRNA-TF 核心调控网络。
本研究通过公共数据库的综合分析,确定了在牙周炎中可能发挥关键作用的特定 TF、miRNA 和 mRNAs。在此基础上,建立了 TF-miRNA-mRNA 网络,为牙周炎调控机制网络提供了全面的视角。