Department of Computational Biology, Carnegie Mellon University, Pittsburgh, PA, USA.
Neuroscience Institute, Carnegie Mellon University, Pittsburgh, PA, USA.
BMC Genomics. 2022 Apr 11;23(1):291. doi: 10.1186/s12864-022-08450-7.
Evolutionary conservation is an invaluable tool for inferring functional significance in the genome, including regions that are crucial across many species and those that have undergone convergent evolution. Computational methods to test for sequence conservation are dominated by algorithms that examine the ability of one or more nucleotides to align across large evolutionary distances. While these nucleotide alignment-based approaches have proven powerful for protein-coding genes and some non-coding elements, they fail to capture conservation of many enhancers, distal regulatory elements that control spatial and temporal patterns of gene expression. The function of enhancers is governed by a complex, often tissue- and cell type-specific code that links combinations of transcription factor binding sites and other regulation-related sequence patterns to regulatory activity. Thus, function of orthologous enhancer regions can be conserved across large evolutionary distances, even when nucleotide turnover is high.
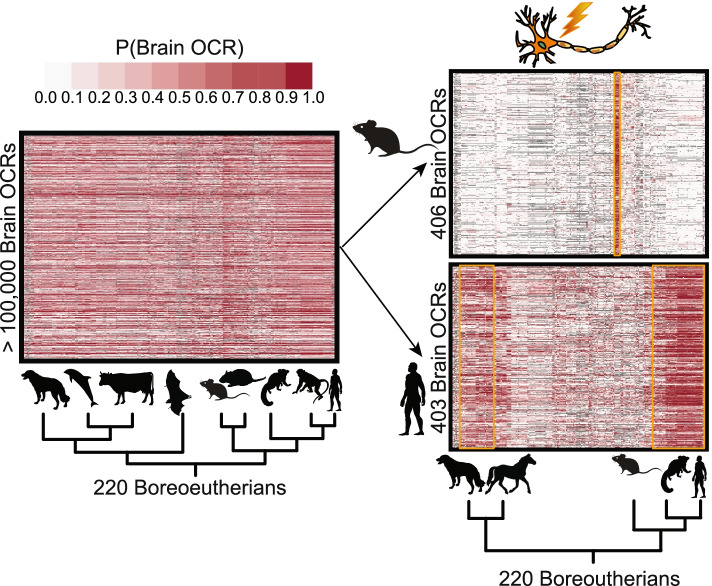
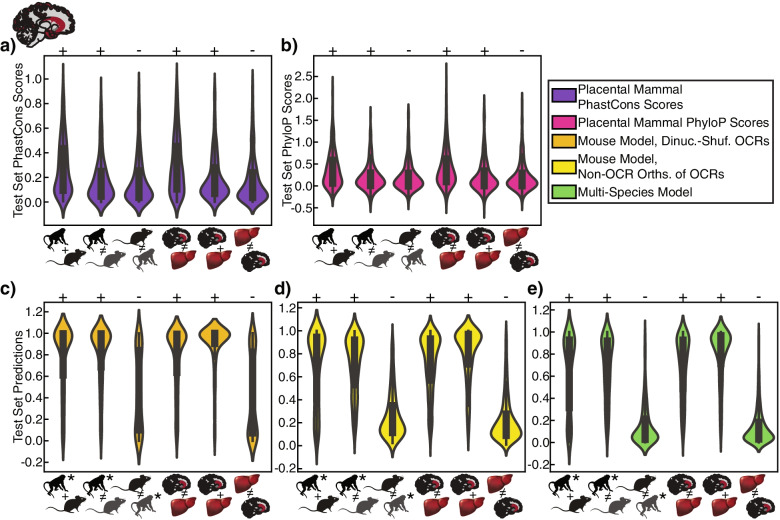
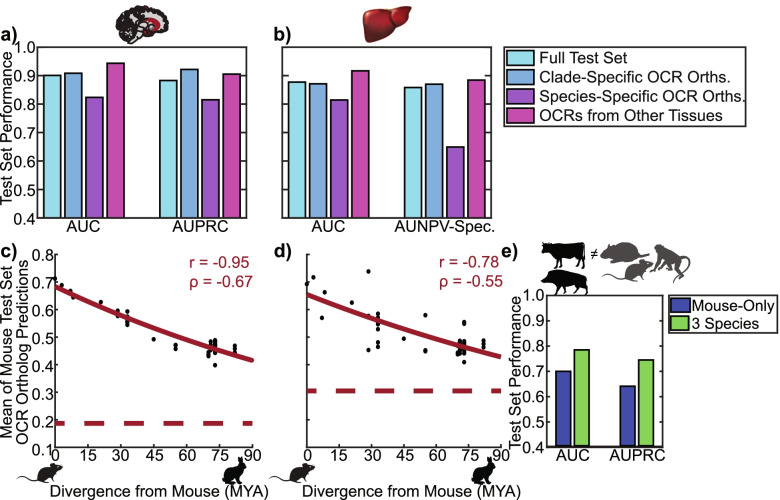
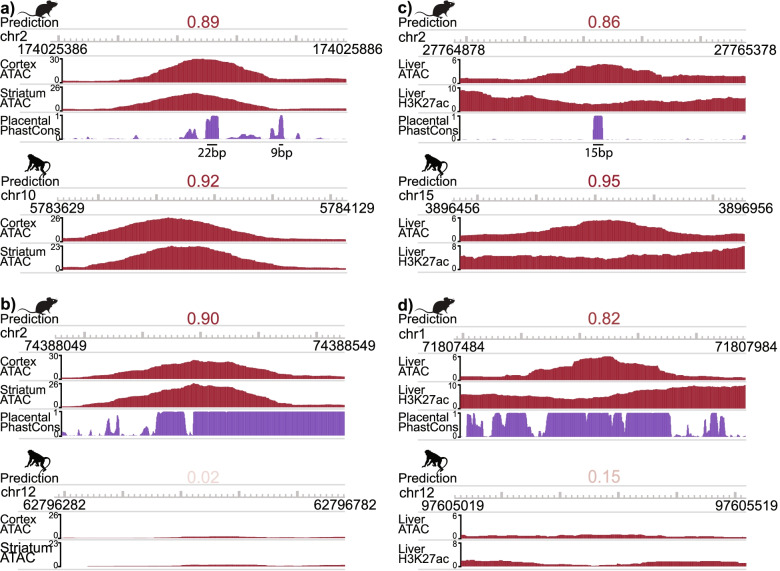
We present a new machine learning-based approach for evaluating enhancer conservation that leverages the combinatorial sequence code of enhancer activity rather than relying on the alignment of individual nucleotides. We first train a convolutional neural network model that can predict tissue-specific open chromatin, a proxy for enhancer activity, across mammals. Next, we apply that model to distinguish instances where the genome sequence would predict conserved function versus a loss of regulatory activity in that tissue. We present criteria for systematically evaluating model performance for this task and use them to demonstrate that our models accurately predict tissue-specific conservation and divergence in open chromatin between primate and rodent species, vastly out-performing leading nucleotide alignment-based approaches. We then apply our models to predict open chromatin at orthologs of brain and liver open chromatin regions across hundreds of mammals and find that brain enhancers associated with neuron activity have a stronger tendency than the general population to have predicted lineage-specific open chromatin.
The framework presented here provides a mechanism to annotate tissue-specific regulatory function across hundreds of genomes and to study enhancer evolution using predicted regulatory differences rather than nucleotide-level conservation measurements.
进化保守性是推断基因组功能意义的宝贵工具,包括在许多物种中至关重要的区域和经历趋同进化的区域。用于测试序列保守性的计算方法主要由算法主导,这些算法检查一个或多个核苷酸在大进化距离上对齐的能力。虽然这些基于核苷酸对齐的方法已被证明对蛋白质编码基因和一些非编码元件非常有效,但它们无法捕捉到许多增强子的保守性,增强子是控制基因表达时空模式的远端调控元件。增强子的功能受一种复杂的、通常是组织和细胞类型特异性的调控,它将转录因子结合位点和其他与调控相关的序列模式的组合与调控活性联系起来。因此,即使核苷酸更替率很高,同源增强子区域的功能也可以在大的进化距离上保守。
我们提出了一种新的基于机器学习的评估增强子保守性的方法,该方法利用增强子活性的组合序列代码,而不是依赖于单个核苷酸的对齐。我们首先训练一个卷积神经网络模型,该模型可以预测哺乳动物中组织特异性的开放染色质,这是增强子活性的一个代理。接下来,我们应用该模型来区分基因组序列预测的保守功能与该组织中失去调控活性的情况。我们提出了用于系统评估该任务的模型性能的标准,并使用它们证明我们的模型可以准确地预测灵长类和啮齿类动物之间组织特异性的开放染色质的保守性和分化,远远超过了领先的基于核苷酸对齐的方法。然后,我们将我们的模型应用于预测数百种哺乳动物中脑和肝开放染色质区域的同源开放染色质,发现与神经元活动相关的脑增强子比一般群体更倾向于具有预测的谱系特异性开放染色质。
这里提出的框架提供了一种机制,可以在数百个基因组中注释组织特异性的调控功能,并使用预测的调控差异而不是核苷酸水平的保守性测量来研究增强子进化。