Mallik Saurav, Zhao Zhongming
Department of Computer Science & Engineering, Aliah University, Newtown, WB-700156, India.
Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston, Houston, TX 77030, USA.
Genes (Basel). 2017 Dec 28;9(1):7. doi: 10.3390/genes9010007.
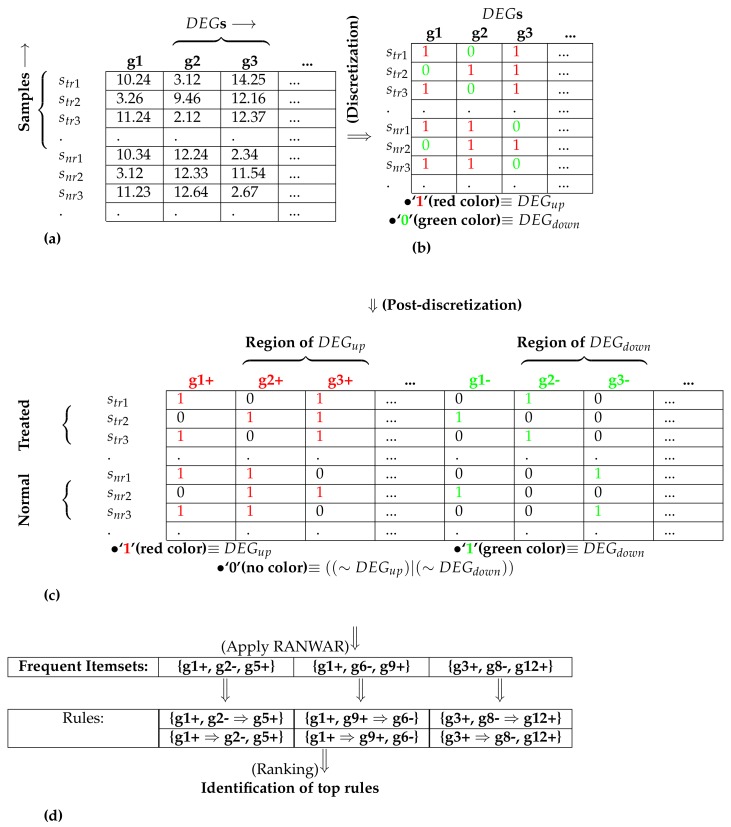
For transcriptomic analysis, there are numerous microarray-based genomic data, especially those generated for cancer research. The typical analysis measures the difference between a cancer sample-group and a matched control group for each transcript or gene. Association rule mining is used to discover interesting item sets through rule-based methodology. Thus, it has advantages to find causal effect relationships between the transcripts. In this work, we introduce two new rule-based similarity measures-weighted rank-based Jaccard and Cosine measures-and then propose a novel computational framework to detect condensed gene co-expression modules ( C o n G E M s) through the association rule-based learning system and the weighted similarity scores. In practice, the list of evolved condensed markers that consists of both singular and complex markers in nature depends on the corresponding condensed gene sets in either antecedent or consequent of the rules of the resultant modules. In our evaluation, these markers could be supported by literature evidence, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway and Gene Ontology annotations. Specifically, we preliminarily identified differentially expressed genes using an empirical Bayes test. A recently developed algorithm-RANWAR-was then utilized to determine the association rules from these genes. Based on that, we computed the integrated similarity scores of these rule-based similarity measures between each rule-pair, and the resultant scores were used for clustering to identify the co-expressed rule-modules. We applied our method to a gene expression dataset for lung squamous cell carcinoma and a genome methylation dataset for uterine cervical carcinogenesis. Our proposed module discovery method produced better results than the traditional gene-module discovery measures. In summary, our proposed rule-based method is useful for exploring biomarker modules from transcriptomic data.
对于转录组分析,有大量基于微阵列的基因组数据,尤其是那些为癌症研究生成的数据。典型的分析是测量每个转录本或基因在癌症样本组和匹配的对照组之间的差异。关联规则挖掘用于通过基于规则的方法发现有趣的项目集。因此,它在发现转录本之间的因果关系方面具有优势。在这项工作中,我们引入了两种新的基于规则的相似性度量——加权基于秩的杰卡德度量和余弦度量——然后提出了一种新颖的计算框架,通过基于关联规则的学习系统和加权相似性分数来检测浓缩基因共表达模块(ConGEMs)。在实践中,由自然状态下的单一和复杂标记组成的进化浓缩标记列表取决于所得模块规则的前件或后件中的相应浓缩基因集。在我们的评估中,这些标记可以得到文献证据、KEGG(京都基因与基因组百科全书)通路和基因本体注释的支持。具体来说,我们使用经验贝叶斯检验初步鉴定差异表达基因。然后利用一种最近开发的算法——RANWAR——从这些基因中确定关联规则。在此基础上,我们计算了每个规则对之间这些基于规则的相似性度量的综合相似性分数,并将所得分数用于聚类以识别共表达规则模块。我们将我们的方法应用于肺鳞状细胞癌的基因表达数据集和子宫颈癌发生的基因组甲基化数据集。我们提出的模块发现方法比传统的基因模块发现度量产生了更好的结果。总之,我们提出的基于规则的方法对于从转录组数据中探索生物标志物模块很有用。