Division of STD Prevention, Centers for Disease Control and Preventiongrid.416738.f, Atlanta, Georgia, USA.
Division of Viral Hepatitis, Centers for Disease Control and Preventiongrid.416738.f, Atlanta, Georgia, USA.
mSphere. 2022 Jun 29;7(3):e0000922. doi: 10.1128/msphere.00009-22. Epub 2022 May 2.
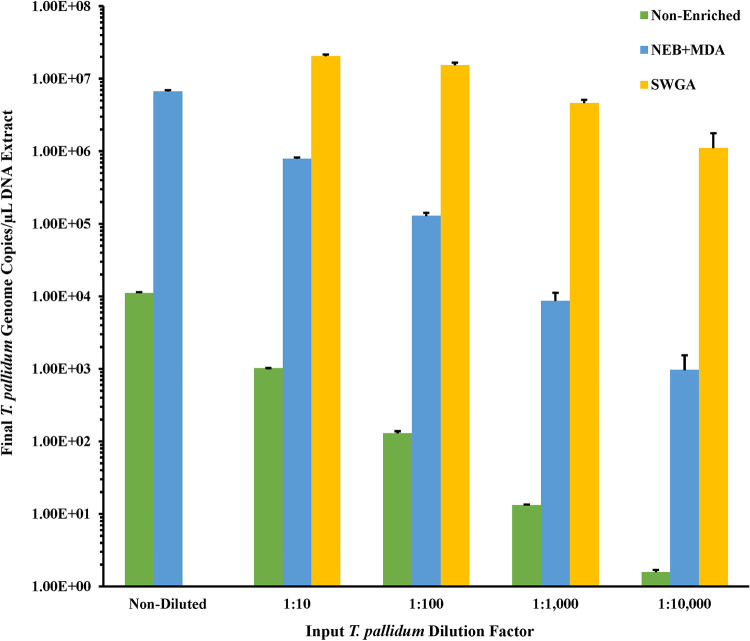
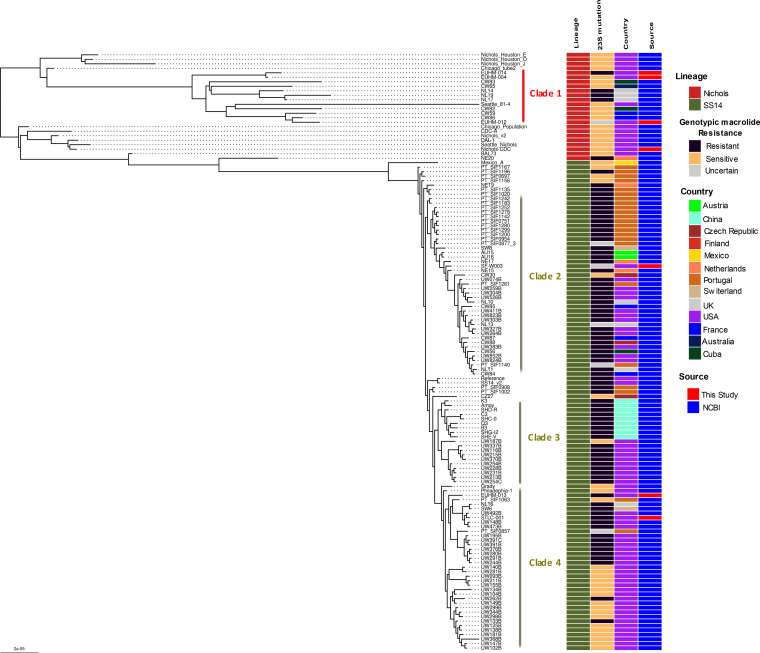
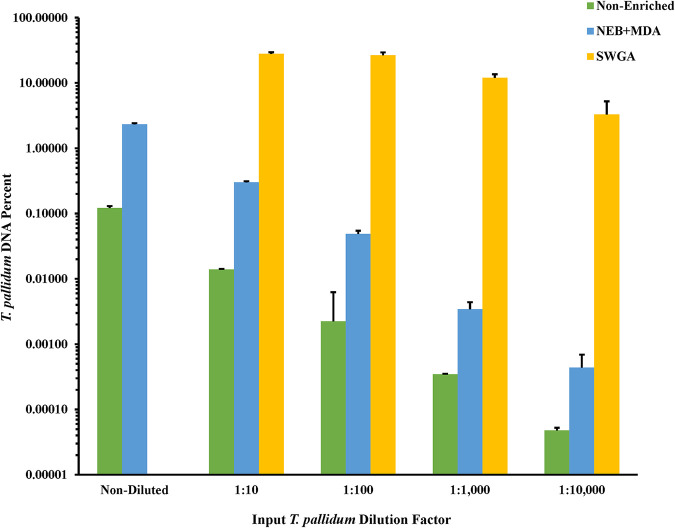
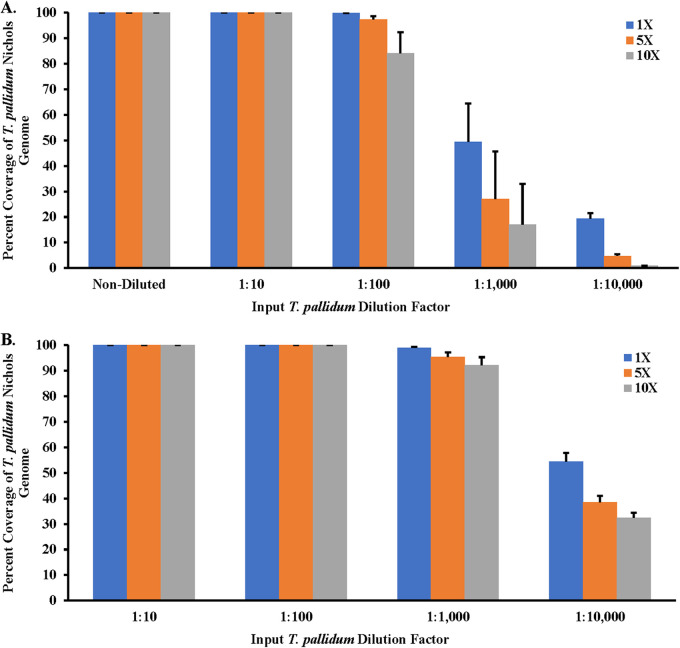
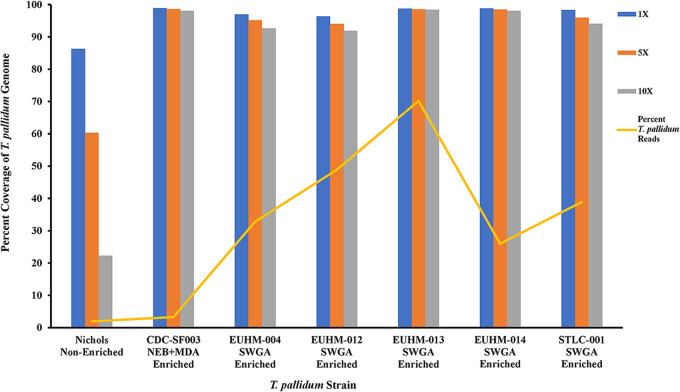
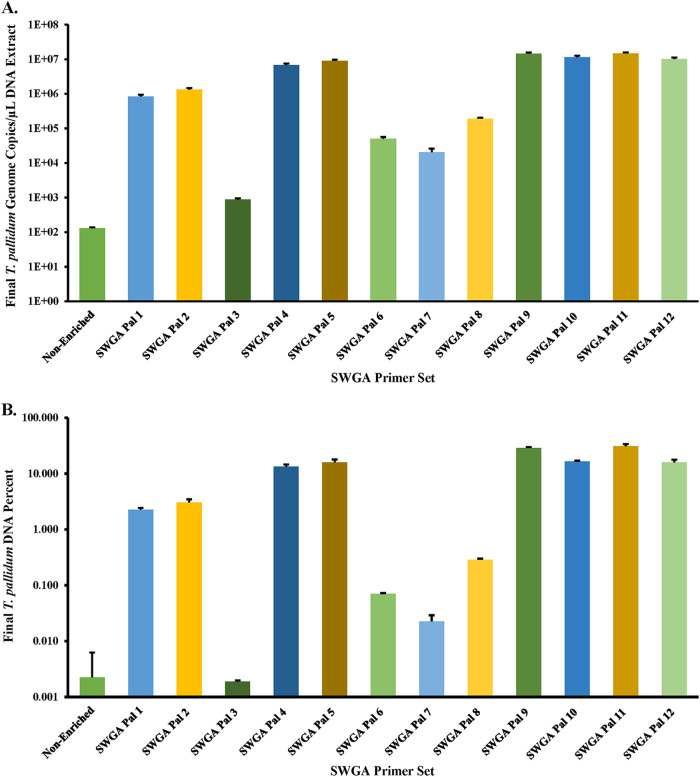
Downstream next-generation sequencing (NGS) of the syphilis spirochete Treponema pallidum subspecies (T. pallidum) is hindered by low bacterial loads and the overwhelming presence of background metagenomic DNA in clinical specimens. In this study, we investigated selective whole-genome amplification (SWGA) utilizing multiple displacement amplification (MDA) in conjunction with custom oligonucleotides with an increased specificity for the T. pallidum genome and the capture and removal of 5'-C-phosphate-G-3' (CpG) methylated host DNA using the NEBNext Microbiome DNA enrichment kit followed by MDA with the REPLI-g single cell kit as enrichment methods to improve the yields of T. pallidum DNA in isolates and lesion specimens from syphilis patients. Sequencing was performed using the Illumina MiSeq v2 500 cycle or NovaSeq 6000 SP platform. These two enrichment methods led to 93 to 98% genome coverage at 5 reads/site in 5 clinical specimens from the United States and rabbit-propagated isolates, containing >14 T. pallidum genomic copies/μL of sample for SWGA and >129 genomic copies/μL for CpG methylation capture with MDA. Variant analysis using sequencing data derived from SWGA-enriched specimens showed that all 5 clinical strains had the A2058G mutation associated with azithromycin resistance. SWGA is a robust method that allows direct whole-genome sequencing (WGS) of specimens containing very low numbers of T. pallidum, which has been challenging until now. Syphilis is a sexually transmitted, disseminated acute and chronic infection caused by the bacterial pathogen Treponema pallidum subspecies . Primary syphilis typically presents as single or multiple mucocutaneous lesions and, if left untreated, can progress through multiple stages with various clinical manifestations. Molecular studies often rely on direct amplification of DNA sequences from clinical specimens; however, this can be impacted by inadequate samples due to disease progression or timing of patients seeking clinical care. While genotyping has provided important data on circulating strains over the past 2 decades, WGS data are needed to better understand strain diversity, perform evolutionary tracing, and monitor antimicrobial resistance markers. The significance of our research is the development of an SWGA DNA enrichment method that expands the range of clinical specimens that can be directly sequenced to include samples with low numbers of T. pallidum.
梅毒螺旋体苍白亚种(T. pallidum)的下游下一代测序(NGS)受到临床标本中低细菌负荷和压倒性存在背景宏基因组 DNA 的阻碍。在这项研究中,我们研究了利用多重置换扩增(MDA)与具有更高 T. pallidum 基因组特异性的定制寡核苷酸结合使用的选择性全基因组扩增(SWGA),以及使用 NEBNext Microbiome DNA 富集试剂盒捕获和去除 5'-C-磷酸-G-3'(CpG)甲基化宿主 DNA,然后使用 REPLI-g 单细胞试剂盒进行 MDA 作为富集方法,以提高梅毒螺旋体 DNA 在梅毒患者的分离物和病变标本中的产量。测序使用 Illumina MiSeq v2 500 循环或 NovaSeq 6000 SP 平台进行。这两种富集方法导致在 5 个来自美国和兔传播分离物的临床标本中,5 个读取/位点的基因组覆盖率为 93%至 98%,SWGA 的样本中包含 >14 个 T. pallidum 基因组拷贝/μL,MDA 的 CpG 甲基化捕获中包含 >129 个基因组拷贝/μL。使用源自 SWGA 富集标本的测序数据进行的变异分析表明,所有 5 个临床株都具有与阿奇霉素耐药相关的 A2058G 突变。SWGA 是一种强大的方法,允许直接对包含极低数量 T. pallidum 的标本进行全基因组测序(WGS),这在以前是具有挑战性的。梅毒是一种由细菌病原体梅毒螺旋体苍白亚种引起的性传播、播散性急性和慢性感染。原发性梅毒通常表现为单个或多个粘膜皮肤损伤,如果未经治疗,可能会通过具有各种临床表现的多个阶段进展。分子研究通常依赖于直接扩增临床标本中的 DNA 序列;然而,由于疾病进展或患者寻求临床护理的时间,这可能会受到样本不足的影响。虽然基因分型在过去 20 年中提供了有关循环菌株的重要数据,但需要 WGS 数据来更好地了解菌株多样性、进行进化追踪和监测抗微生物药物耐药性标记物。我们研究的意义在于开发了一种 SWGA DNA 富集方法,该方法扩大了可以直接测序的临床标本范围,包括 T. pallidum 数量较少的标本。