CBIO, MINES ParisTech, PSL Research University, 75006, Paris, France.
Translational Sciences, SANOFI R&D, 91385, Chilly-Mazarin, France.
BMC Med Genomics. 2022 Apr 30;15(1):100. doi: 10.1186/s12920-022-01247-3.
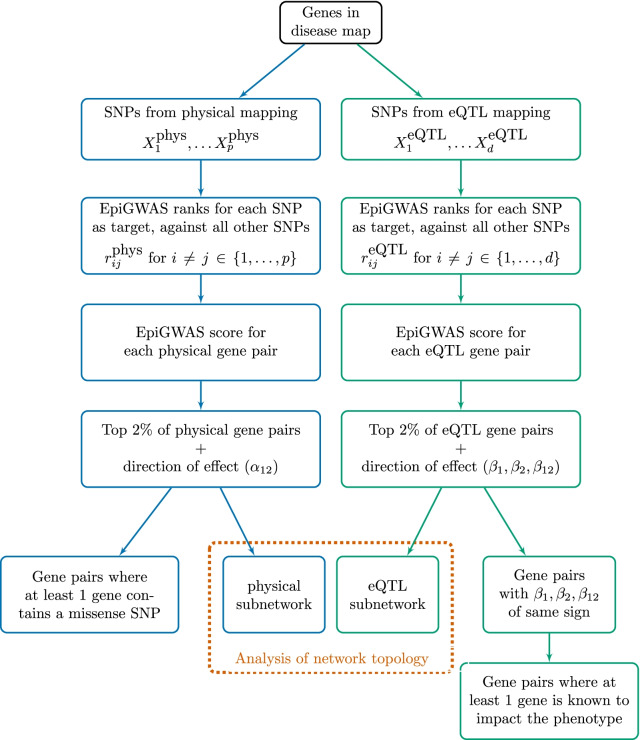
For the most part, genome-wide association studies (GWAS) have only partially explained the heritability of complex diseases. One of their limitations is to assume independent contributions of individual variants to the phenotype. Many tools have therefore been developed to investigate the interactions between distant loci, or epistasis. Among them, the recently proposed EpiGWAS models the interactions between a target variant and the rest of the genome. However, applying this approach to studying interactions along all genes of a disease map is not straightforward. Here, we propose a pipeline to that effect, which we illustrate by investigating a multiple sclerosis GWAS dataset from the Wellcome Trust Case Control Consortium 2 through 19 disease maps from the MetaCore pathway database.
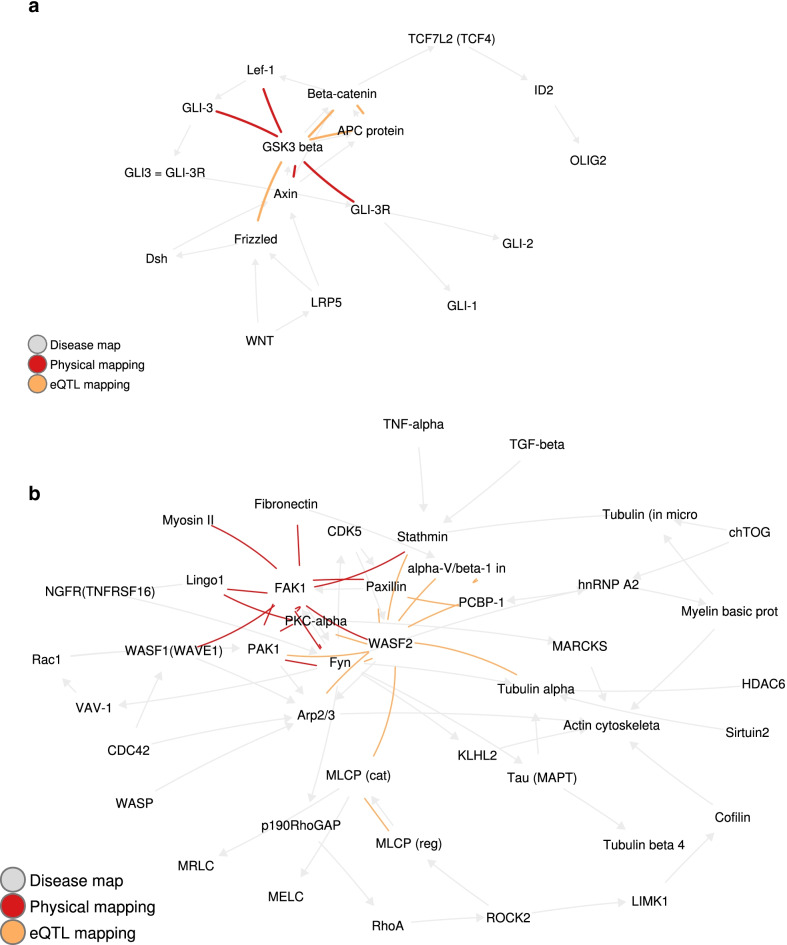
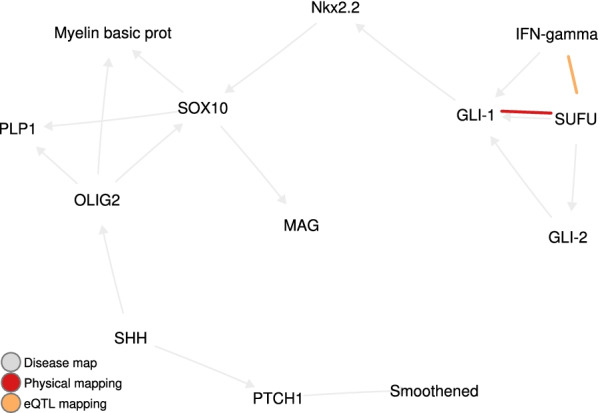
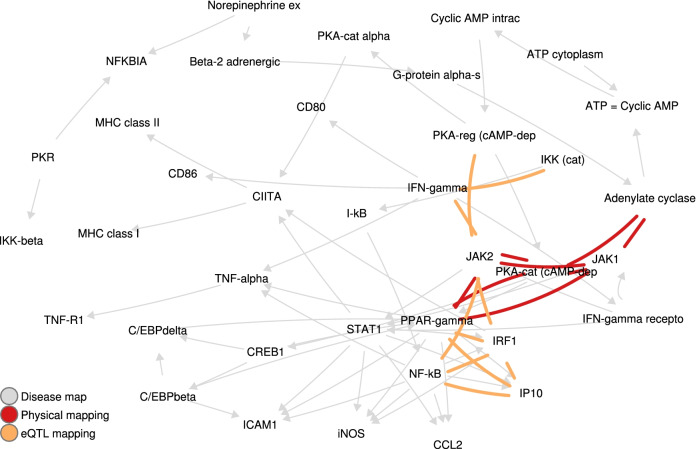
For each disease map, we build an epistatic network by connecting the genes that are deemed to interact. These networks tend to be connected, complementary to the disease maps and contain hubs. In addition, we report 4 epistatic gene pairs involving missense variants, and 25 gene pairs with a deleterious epistatic effect mediated by eQTLs. Among these, we highlight the interaction of GLI-1 and SUFU, and of IP10 and NF-[Formula: see text]B, as they both match known biological interactions. The latter pair is particularly promising for therapeutic development, as both genes have known inhibitors.
Our study showcases the ability of EpiGWAS to uncover biologically interpretable epistatic interactions that are potentially actionable for the development of combination therapy.
大多数情况下,全基因组关联研究(GWAS)仅部分解释了复杂疾病的遗传率。其局限性之一是假设个体变异对表型的独立贡献。因此,已经开发了许多工具来研究远隔基因座之间的相互作用,或上位性。其中,最近提出的 EpiGWAS 模型可以模拟目标变体与基因组其余部分之间的相互作用。然而,直接将这种方法应用于研究疾病图谱中所有基因的相互作用并不容易。在这里,我们提出了一种实现这一目标的方法,并通过研究来自 Wellcome Trust Case Control Consortium 2 的多发性硬化症 GWAS 数据集以及来自 MetaCore 途径数据库的 19 个疾病图谱来说明。
对于每个疾病图谱,我们通过连接被认为相互作用的基因来构建上位性网络。这些网络往往是相互连接的,与疾病图谱互补,并且包含枢纽。此外,我们报告了涉及错义变异的 4 对上位性基因对,以及涉及 eQTLs 的 25 对具有有害上位性效应的基因对。其中,我们强调了 GLI-1 和 SUFU 以及 IP10 和 NF-[Formula: see text]B 之间的相互作用,因为它们都与已知的生物学相互作用相匹配。后者对治疗开发特别有希望,因为这两个基因都有已知的抑制剂。
我们的研究展示了 EpiGWAS 发现潜在可用于开发联合治疗的生物学可解释上位性相互作用的能力。