Genomic Epidemiology Branch, International Agency for Research on Cancer/World Health Organization (IARC/WHO), Lyon, France.
Cancer Epidemiology Unit, Nuffield Department of Population Health, University of Oxford, Oxford, England.
J Natl Cancer Inst. 2022 Aug 8;114(8):1159-1166. doi: 10.1093/jnci/djac087.
Germline genetic variation contributes to lung cancer (LC) susceptibility. Previous genome-wide association studies (GWAS) have implicated susceptibility loci involved in smoking behaviors and DNA repair genes, but further work is required to identify susceptibility variants.
To identify LC susceptibility loci, a family history-based genome-wide association by proxy (GWAx) of LC (48 843 European proxy LC patients, 195 387 controls) was combined with a previous LC GWAS (29 266 patients, 56 450 controls) by meta-analysis. Colocalization was used to explore candidate genes and overlap with existing traits at discovered susceptibility loci. Polygenic risk scores (PRS) were tested within an independent validation cohort (1 666 LC patients vs 6 664 controls) using variants selected from the LC susceptibility loci and a novel selection approach using published GWAS summary statistics. Finally, the effects of the LC PRS on somatic mutational burden were explored in patients whose tumor resections have been profiled by exome (n = 685) and genome sequencing (n = 61). Statistical tests were 2-sided.
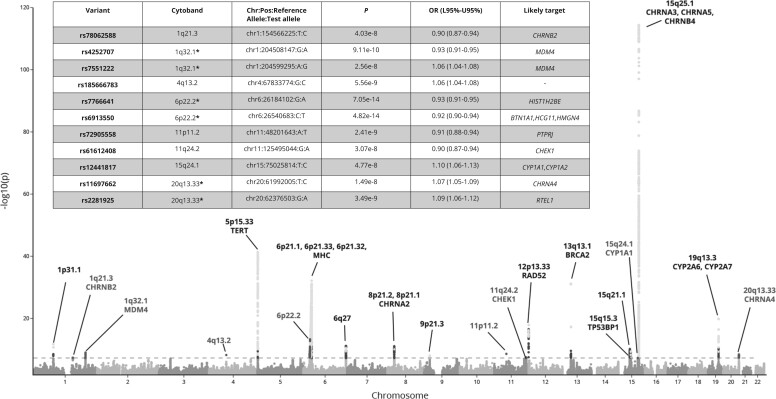
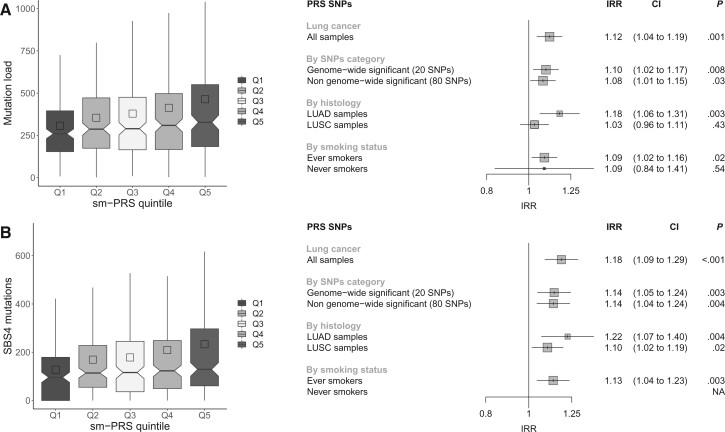
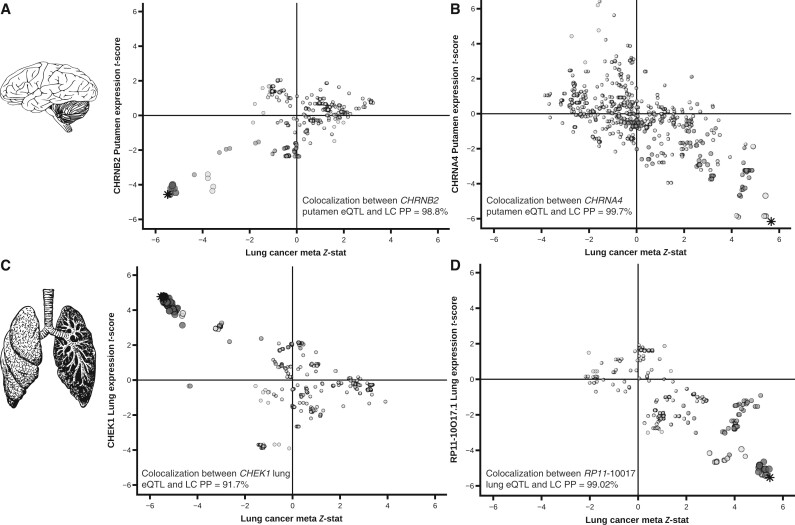
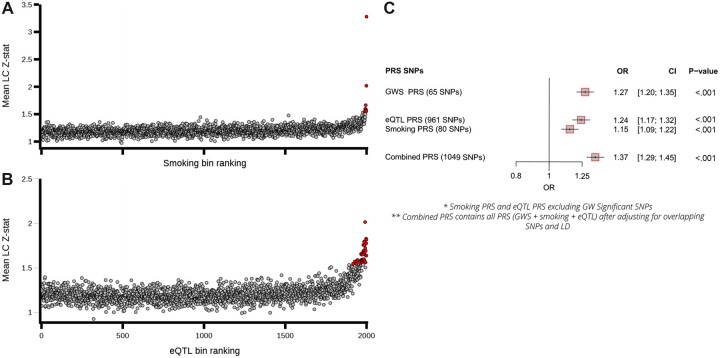
The GWAx-GWAS meta-analysis identified 8 novel LC loci. Colocalization implicated DNA repair genes (CHEK1), metabolic genes (CYP1A1), and smoking propensity genes (CHRNA4 and CHRNB2). PRS analysis demonstrated that these variants, as well as subgenome-wide significant variants related to expression quantitative trait loci and/or smoking propensity, assisted in LC genetic risk prediction (odds ratio = 1.37, 95% confidence interval = 1.29 to 1.45; P < .001). Patients with higher genetic PRS loads of smoking-related variants tended to have higher mutation burdens in their lung tumors.
This study has expanded the number of LC susceptibility loci and provided insights into the molecular mechanisms by which these susceptibility variants contribute to LC development.
种系遗传变异可导致肺癌(LC)易感性。先前的全基因组关联研究(GWAS)已经发现了与吸烟行为和 DNA 修复基因相关的易感基因座,但仍需要进一步工作来鉴定易感变异。
为了鉴定 LC 易感性基因座,通过代理进行基于家族史的全基因组关联(GWAx)(48843 名欧洲代理 LC 患者,195387 名对照),并与先前的 LC GWAS(29266 名患者,56450 名对照)进行荟萃分析。共定位用于在发现的易感基因座中探索候选基因,并与现有性状重叠。多基因风险评分(PRS)在独立验证队列中进行测试(1666 名 LC 患者与 6664 名对照),使用从 LC 易感性基因座中选择的变体和使用已发表的 GWAS 汇总统计数据的新选择方法。最后,在经过外显子组(n=685)和基因组测序(n=61)分析的患者中,探索了 LC PRS 对体细胞突变负担的影响。统计检验为双侧。
GWAx-GWAS 荟萃分析确定了 8 个新的 LC 基因座。共定位表明 DNA 修复基因(CHEK1)、代谢基因(CYP1A1)和吸烟倾向基因(CHRNA4 和 CHRNB2)。PRS 分析表明,这些变体以及与表达数量性状基因座和/或吸烟倾向相关的亚基因组全基因组显著变体有助于 LC 遗传风险预测(优势比=1.37,95%置信区间=1.29 至 1.45;P<0.001)。具有更高的与吸烟相关变体的遗传 PRS 负荷的患者,其肺部肿瘤的突变负担往往更高。
本研究扩大了 LC 易感基因座的数量,并深入了解了这些易感变异导致 LC 发展的分子机制。