Pandeswari P Boomathi, Sabareesh Varatharajan
Advanced Centre for Bio Separation Technology (CBST), Vellore Institute of Technology (VIT) Vellore Tamil Nadu 632014 India
RSC Adv. 2019 Jan 2;9(1):313-344. doi: 10.1039/c8ra07200k. eCollection 2018 Dec 19.
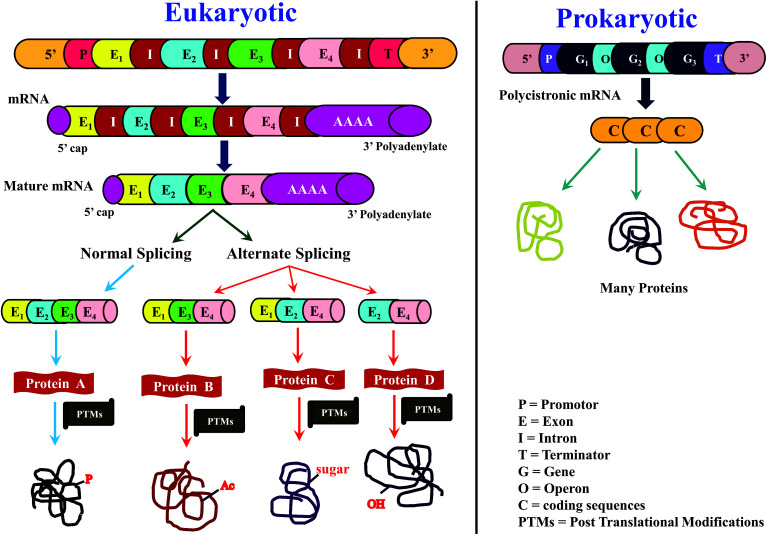
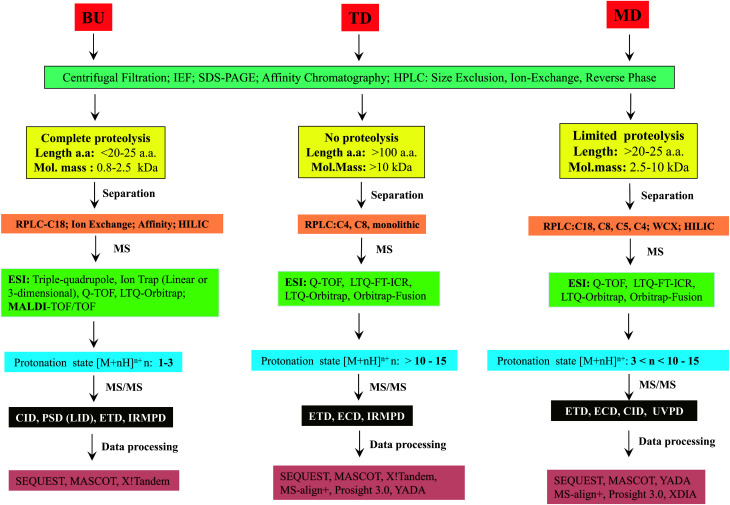
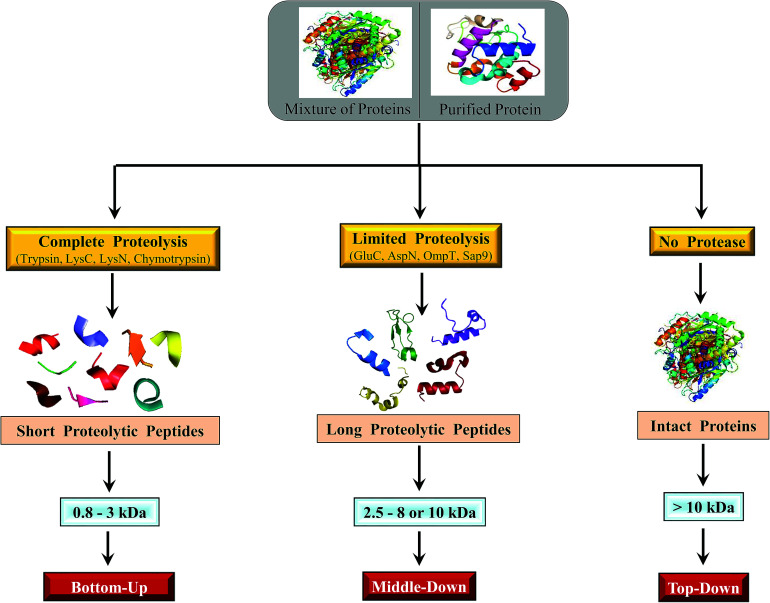
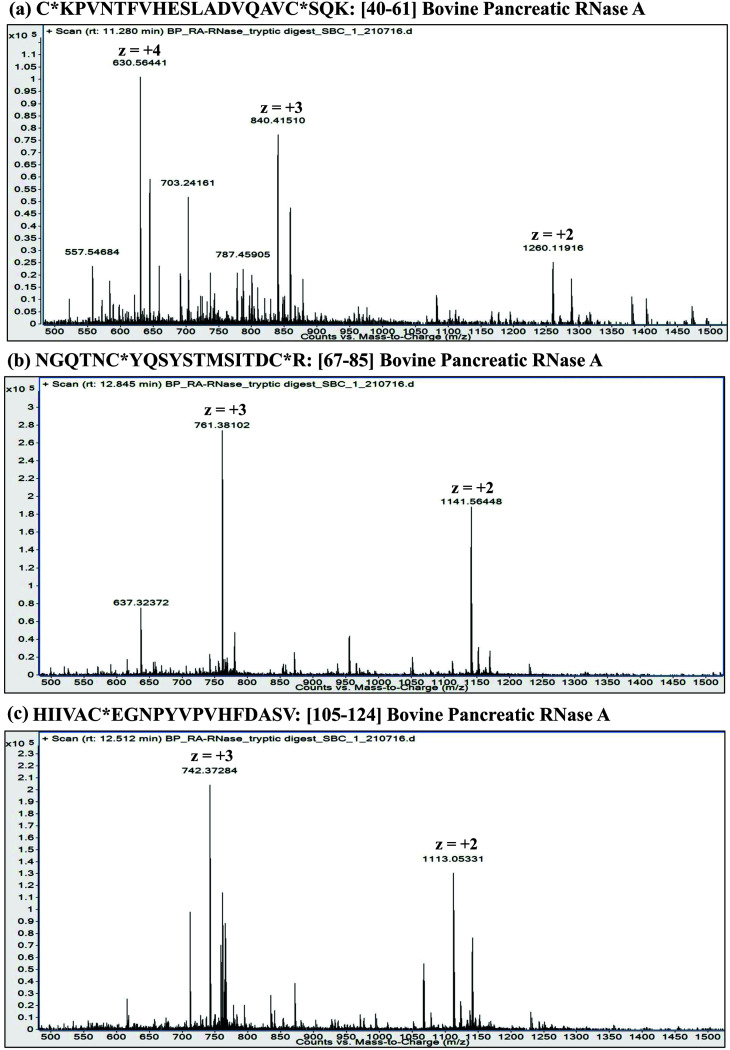
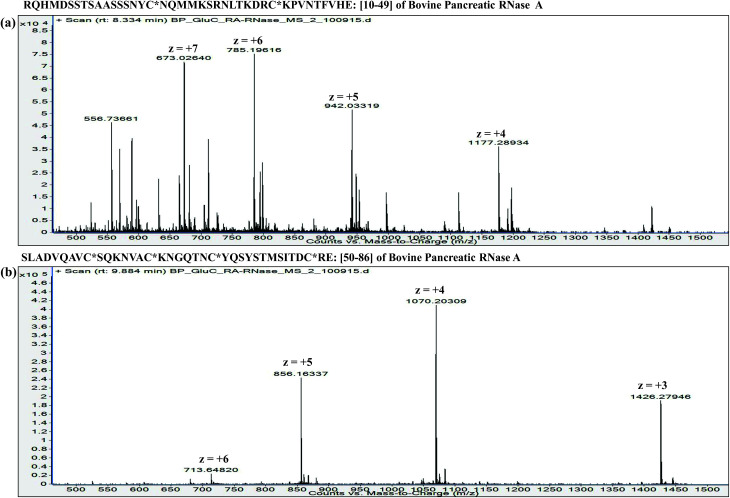
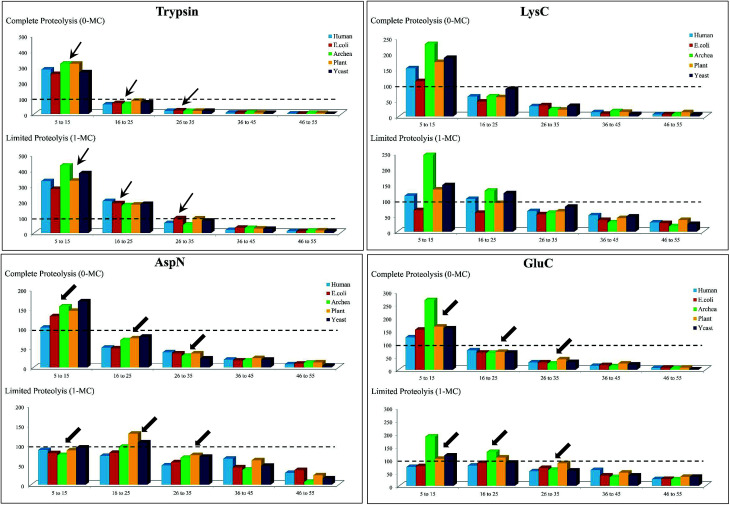
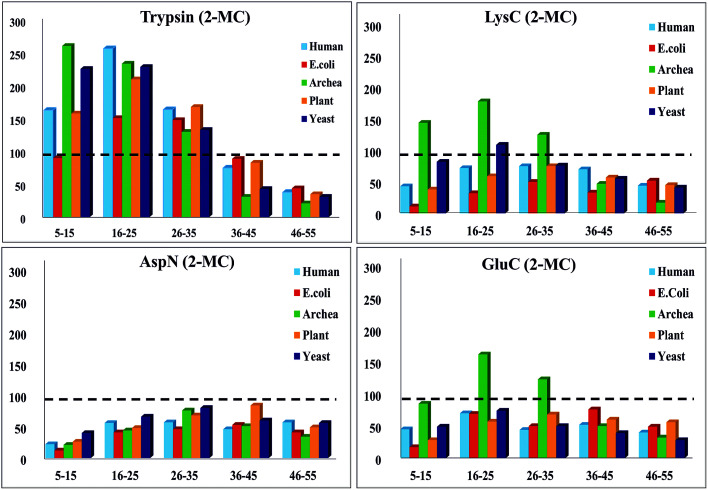
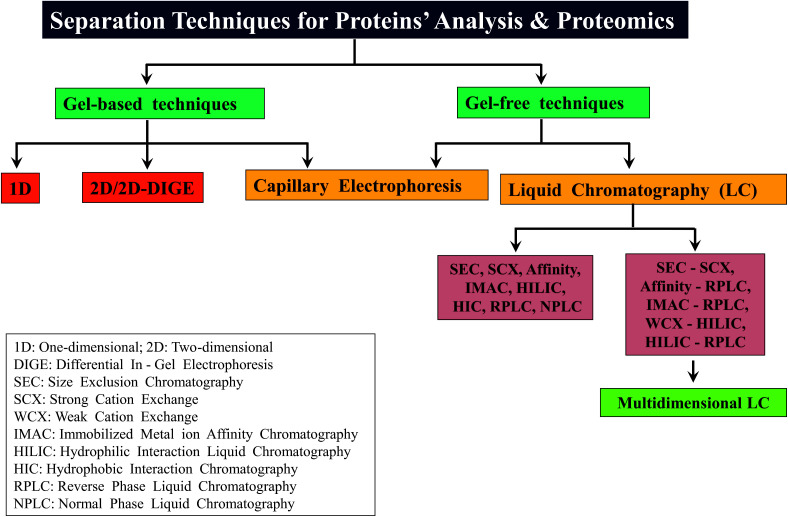
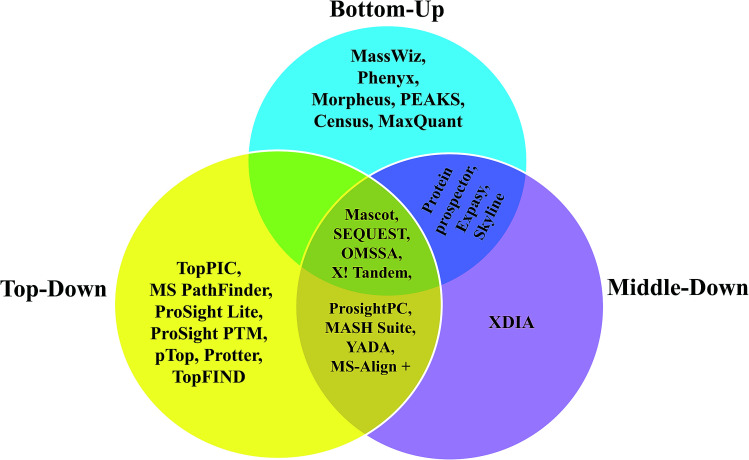
Owing to rapid growth in the elucidation of genome sequences of various organisms, deducing proteome sequences has become imperative, in order to have an improved understanding of biological processes. Since the traditional Edman method was unsuitable for high-throughput sequencing and also for N-terminus modified proteins, mass spectrometry (MS) based methods, mainly based on soft ionization modes: electrospray ionization and matrix-assisted laser desorption/ionization, began to gain significance. MS based methods were adaptable for high-throughput studies and applicable for sequencing N-terminus blocked proteins/peptides too. Consequently, over the last decade a new discipline called 'proteomics' has emerged, which encompasses the attributes necessary for high-throughput identification of proteins. 'Proteomics' may also be regarded as an offshoot of the classic field, 'biochemistry'. Many protein sequencing and proteomic investigations were successfully accomplished through MS dependent sequence elucidation of 'short proteolytic peptides (typically: 7-20 amino acid residues), which is called the 'shotgun' or 'bottom-up (BU)' approach. While the BU approach continues as a workhorse for proteomics/protein sequencing, attempts to sequence intact proteins without proteolysis, called the 'top-down (TD)' approach started, due to ambiguities in the BU approach, , protein inference problem, identification of proteoforms and the discovery of posttranslational modifications (PTMs). The high-throughput TD approach (TD proteomics) is yet in its infancy. Nevertheless, TD characterization of purified intact proteins has been useful for detecting PTMs. With the hope to overcome the pitfalls of BU and TD strategies, another concept called the 'middle-down (MD)' approach was put forward. Similar to BU, the MD approach also involves proteolysis, but in a restricted manner, to produce 'longer' proteolytic peptides than the ones usually obtained in BU studies, thereby providing better sequence coverage. In this regard, special proteases (OmpT, Sap9, IdeS) have been used, which can cleave proteins to produce longer proteolytic peptides. By reviewing ample evidences currently existing in the literature that is predominantly on PTM characterization of histones and antibodies, herein we highlight salient features of the MD approach. Consequently, we are inclined to claim that the MD concept might have widespread applications in future for various research areas, such as clinical, biopharmaceuticals (including PTM analysis) and even for general/routine characterization of proteins including therapeutic proteins, but not just limited to analysis of histones or antibodies.
由于各种生物体基因组序列的解析迅速发展,为了更好地理解生物过程,推导蛋白质组序列变得势在必行。由于传统的埃德曼方法不适用于高通量测序,也不适用于N端修饰的蛋白质,基于质谱(MS)的方法开始变得重要起来,这些方法主要基于软电离模式:电喷雾电离和基质辅助激光解吸/电离。基于MS的方法适用于高通量研究,也适用于对N端封闭的蛋白质/肽进行测序。因此,在过去十年中出现了一门名为“蛋白质组学”的新学科,它涵盖了高通量鉴定蛋白质所需的特性。“蛋白质组学”也可被视为经典领域“生物化学”的一个分支。许多蛋白质测序和蛋白质组学研究通过对“短蛋白水解肽(通常为:7-20个氨基酸残基)”进行基于MS的序列解析而成功完成,这被称为“鸟枪法”或“自下而上(BU)”方法。虽然BU方法仍然是蛋白质组学/蛋白质测序的主力军,但由于BU方法存在的模糊性、蛋白质推断问题、蛋白变体的鉴定以及翻译后修饰(PTM)的发现,人们开始尝试对完整蛋白质进行无需蛋白水解的测序,即“自上而下(TD)”方法。高通量TD方法(TD蛋白质组学)仍处于起步阶段。然而,对纯化的完整蛋白质进行TD表征对于检测PTM很有用。为了克服BU和TD策略的缺陷,人们提出了另一个概念,即“中而下(MD)”方法。与BU类似,MD方法也涉及蛋白水解,但方式有限,以产生比BU研究中通常获得的“更长”的蛋白水解肽,从而提供更好的序列覆盖。在这方面,人们使用了特殊的蛋白酶(OmpT、Sap9、IdeS),它们可以切割蛋白质以产生更长的蛋白水解肽。通过回顾目前文献中大量主要关于组蛋白和抗体PTM表征的证据,我们在此强调MD方法的显著特征。因此,我们倾向于认为MD概念可能在未来广泛应用于各种研究领域,如临床、生物制药(包括PTM分析),甚至用于包括治疗性蛋白质在内的蛋白质的一般/常规表征,而不仅仅局限于组蛋白或抗体的分析。