SJTU-Yale Joint Center for Biostatistics and Data Science, Department of Bioinformatics and Biostatistics, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China; Center for Biomedical Informatics, Shanghai Engineering Research Center for Big Data in Pediatric Precision Medicine, Shanghai Children's Hospital, Shanghai 200040, China; Department of Biostatistics, Yale University, New Haven, CT 06511, USA.
SJTU-Yale Joint Center for Biostatistics and Data Science, Department of Bioinformatics and Biostatistics, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
Genomics Proteomics Bioinformatics. 2023 Feb;21(1):164-176. doi: 10.1016/j.gpb.2021.08.017. Epub 2022 May 13.
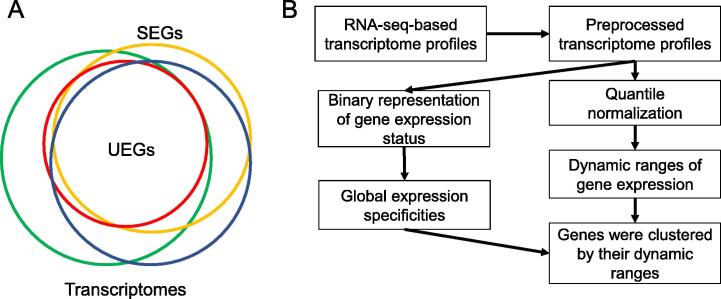
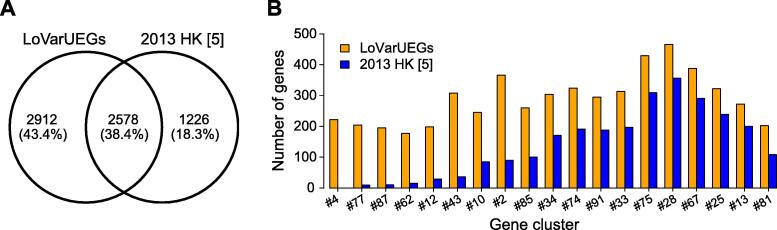
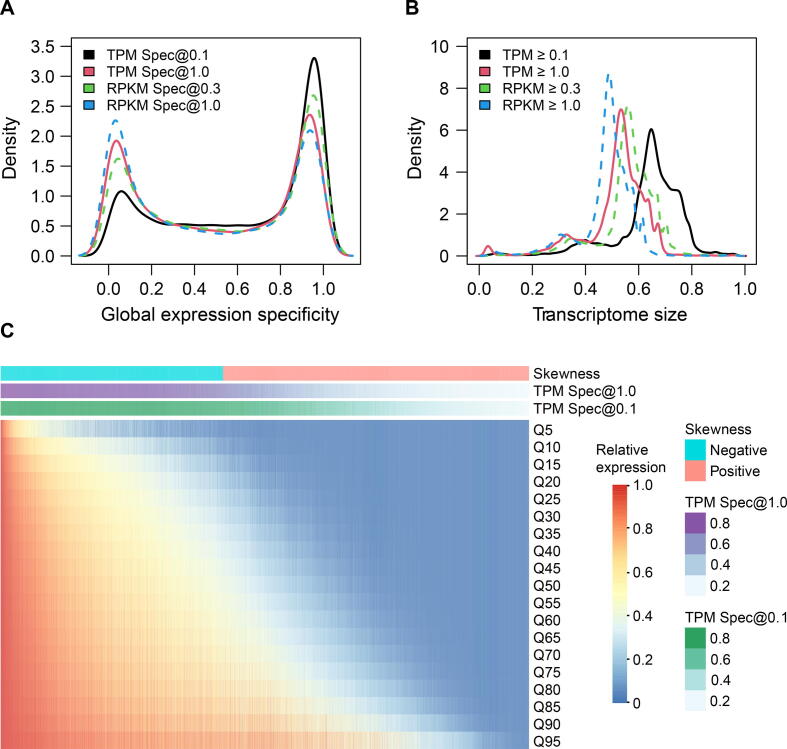
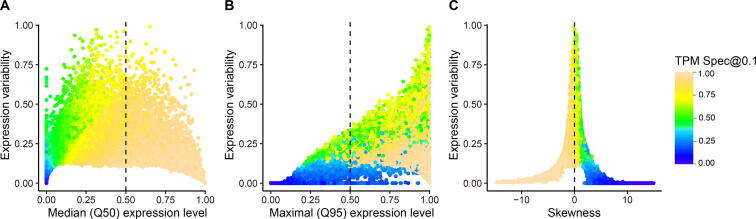
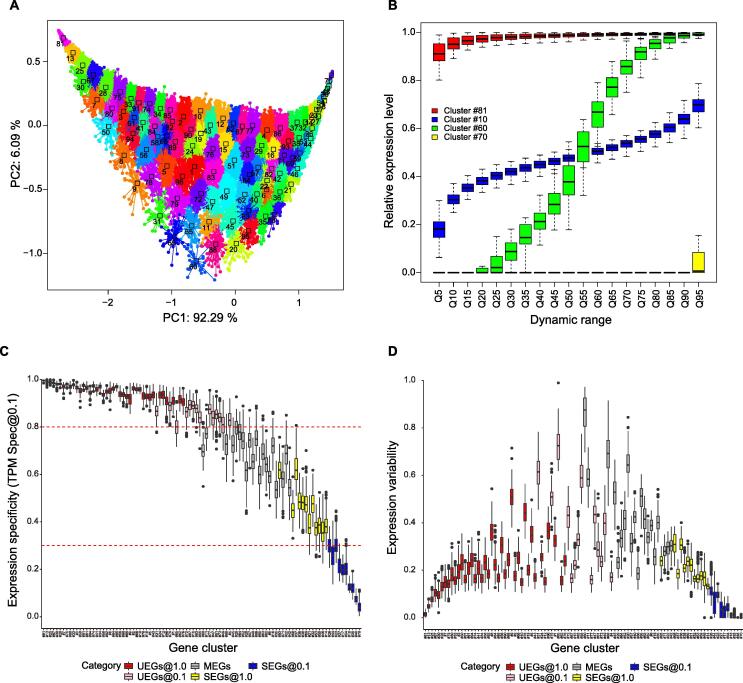
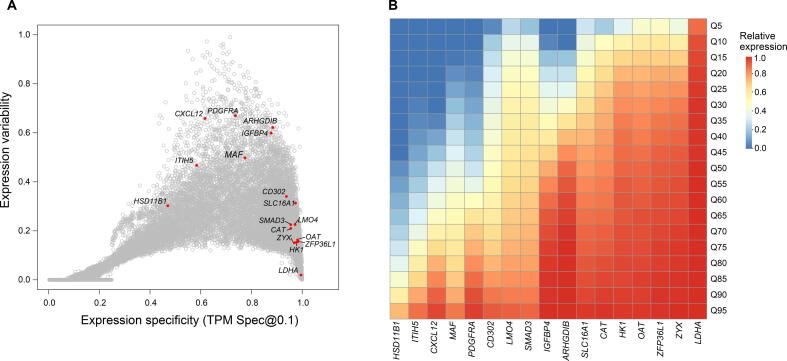
Comprehensive characterization of spatial and temporal gene expression patterns in humans is critical for uncovering the regulatory codes of the human genome and understanding the molecular mechanisms of human diseases. Ubiquitously expressed genes (UEGs) refer to the genes expressed across a majority of, if not all, phenotypic and physiological conditions of an organism. It is known that many human genes are broadly expressed across tissues. However, most previous UEG studies have only focused on providing a list of UEGs without capturing their global expression patterns, thus limiting the potential use of UEG information. In this study, we proposed a novel data-driven framework to leverage the extensive collection of ∼ 40,000 human transcriptomes to derive a list of UEGs and their corresponding global expression patterns, which offers a valuable resource to further characterize human transcriptome. Our results suggest that about half (12,234; 49.01%) of the human genes are expressed in at least 80% of human transcriptomes, and the median size of the human transcriptome is 16,342 genes (65.44%). Through gene clustering, we identified a set of UEGs, named LoVarUEGs, which have stable expression across human transcriptomes and can be used as internal reference genes for expression measurement. To further demonstrate the usefulness of this resource, we evaluated the global expression patterns for 16 previously predicted disallowed genes in islet beta cells and found that seven of these genes showed relatively more varied expression patterns, suggesting that the repression of these genes may not be unique to islet beta cells.
全面描绘人类时空基因表达模式对于揭示人类基因组的调控密码和理解人类疾病的分子机制至关重要。普遍表达基因(UEGs)是指在生物体的大多数(如果不是全部)表型和生理条件下都表达的基因。已知许多人类基因在组织中广泛表达。然而,大多数先前的 UEG 研究仅侧重于提供 UEG 的列表,而没有捕捉到它们的全局表达模式,从而限制了 UEG 信息的潜在用途。在这项研究中,我们提出了一种新颖的数据驱动框架,利用大约 40,000 个人类转录组的广泛收集来得出 UEG 及其相应的全局表达模式列表,这为进一步描述人类转录组提供了有价值的资源。我们的结果表明,约一半(12,234 个;49.01%)的人类基因在至少 80%的人类转录组中表达,人类转录组的中位数大小为 16,342 个基因(65.44%)。通过基因聚类,我们鉴定了一组 UEGs,命名为 LoVarUEGs,它们在人类转录组中具有稳定的表达,可以用作表达测量的内部参考基因。为了进一步证明这个资源的有用性,我们评估了 16 个先前预测的胰岛β细胞中不允许的基因的全局表达模式,发现其中 7 个基因的表达模式相对更为多样化,这表明这些基因的抑制作用可能不是胰岛β细胞所特有的。