Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, MD 21205.
Department of Applied Mathematics and Statistics, Johns Hopkins University, Baltimore, MD 21218.
Proc Natl Acad Sci U S A. 2018 May 1;115(18):4545-4552. doi: 10.1073/pnas.1721628115. Epub 2018 Apr 16.
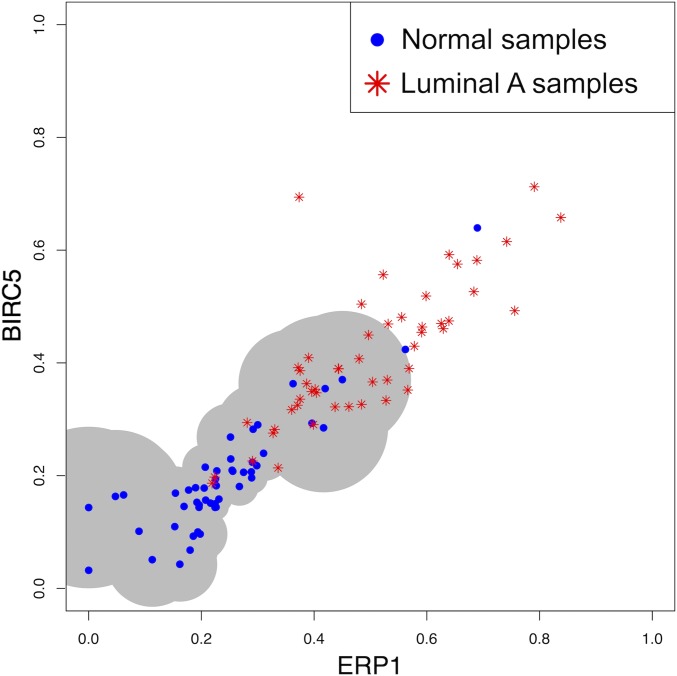
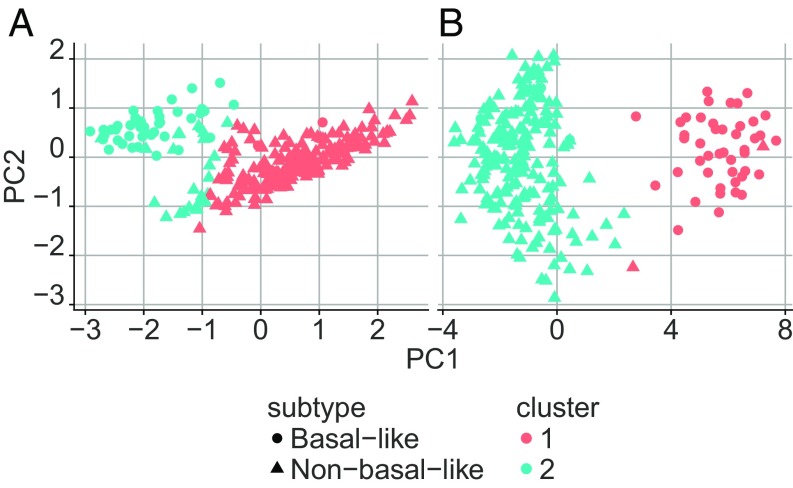
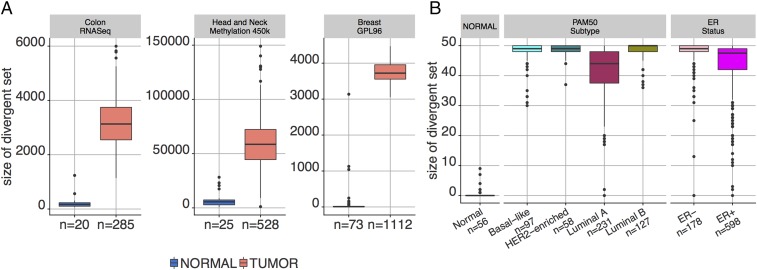
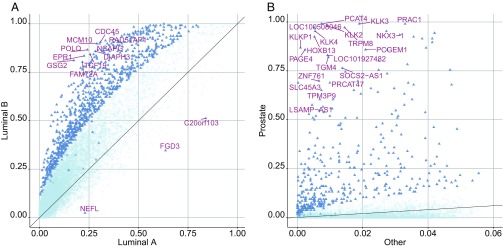
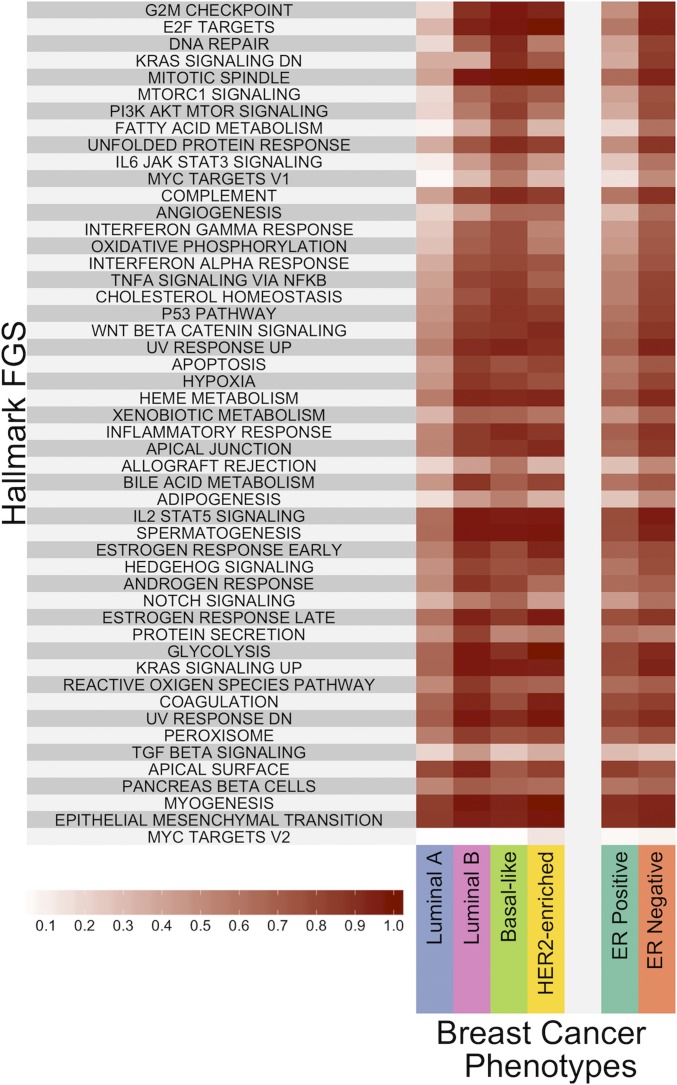
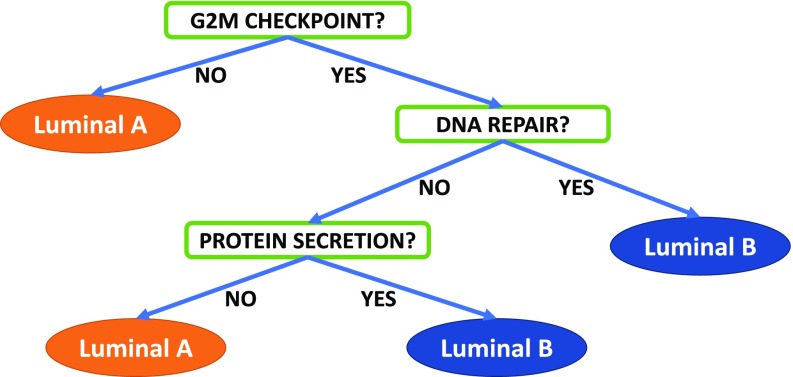
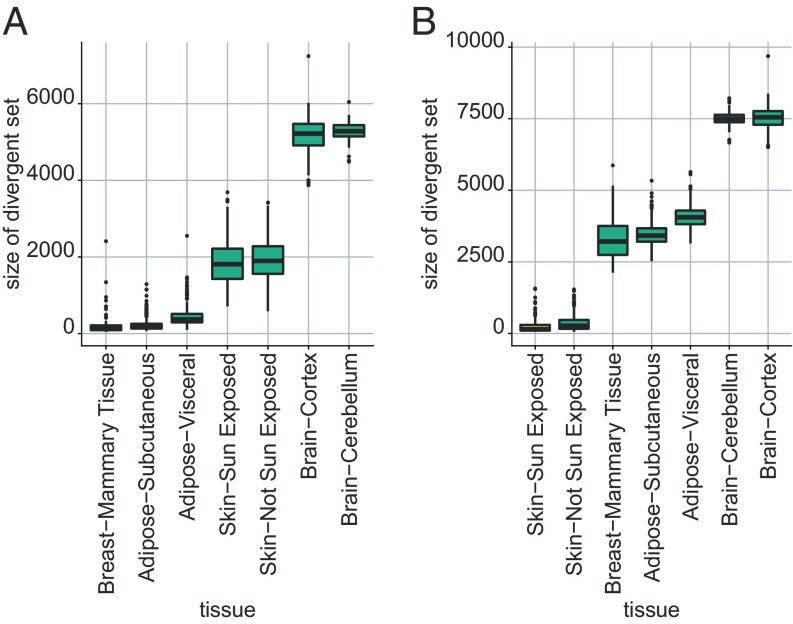
Data collected from omics technologies have revealed pervasive heterogeneity and stochasticity of molecular states within and between phenotypes. A prominent example of such heterogeneity occurs between genome-wide mRNA, microRNA, and methylation profiles from one individual tumor to another, even within a cancer subtype. However, current methods in bioinformatics, such as detecting differentially expressed genes or CpG sites, are population-based and therefore do not effectively model intersample diversity. Here we introduce a unified theory to quantify sample-level heterogeneity that is applicable to a single omics profile. Specifically, we simplify an omics profile to a digital representation based on the omics profiles from a set of samples from a reference or baseline population (e.g., normal tissues). The state of any subprofile (e.g., expression vector for a subset of genes) is said to be "divergent" if it lies outside the estimated support of the baseline distribution and is consequently interpreted as "dysregulated" relative to that baseline. We focus on two cases: single features (e.g., individual genes) and distinguished subsets (e.g., regulatory pathways). Notably, since the divergence analysis is at the individual sample level, dysregulation can be analyzed probabilistically; for example, one can estimate the probability that a gene or pathway is divergent in some population. Finally, the reduction in complexity facilitates a more "personalized" and biologically interpretable analysis of variation, as illustrated by experiments involving tissue characterization, disease detection and progression, and disease-pathway associations.
组学技术所收集的数据揭示了分子状态在表型内和表型之间普遍存在的异质性和随机性。这种异质性的一个突出例子发生在个体肿瘤之间的全基因组 mRNA、microRNA 和甲基化谱之间,即使在癌症亚型内也是如此。然而,生物信息学中的当前方法,如检测差异表达的基因或 CpG 位点,是基于群体的,因此不能有效地模拟样本间的多样性。在这里,我们引入了一种统一的理论来量化样本水平的异质性,该理论适用于单个组学图谱。具体来说,我们将组学图谱简化为基于参考或基线人群(例如正常组织)中一组样本的组学图谱的数字表示。如果任何子图谱(例如,一组基因的表达向量)的状态位于估计的基线分布的支持范围之外,则表示该状态“发散”,并且相对于该基线被解释为“失调”。我们专注于两种情况:单个特征(例如,单个基因)和有区别的子集(例如,调控途径)。值得注意的是,由于发散分析是在单个样本水平上进行的,因此可以对失调进行概率分析;例如,可以估计某个基因或途径在某些人群中发散的概率。最后,通过实验涉及组织特征描述、疾病检测和进展以及疾病途径关联,简化复杂性促进了更“个性化”和生物学可解释的变异分析。