You Jingyi, Li Min, Li Hongwei, Bai Yulin, Zhu Xuan, Kong Xiangjun, Chen Xiaoyan, Zhou Ruiyang
Key Laboratory of Plant Genetics and Breeding, College of Agriculture, Guangxi University, Nanning, China.
Xinjiang Yida Textile Co., Ltd, Urumqi, China.
Front Plant Sci. 2022 Apr 27;13:770098. doi: 10.3389/fpls.2022.770098. eCollection 2022.
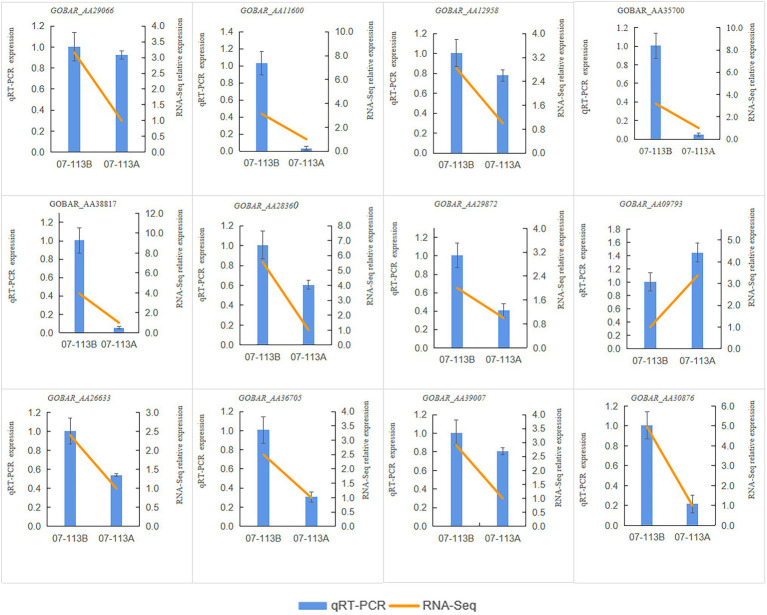
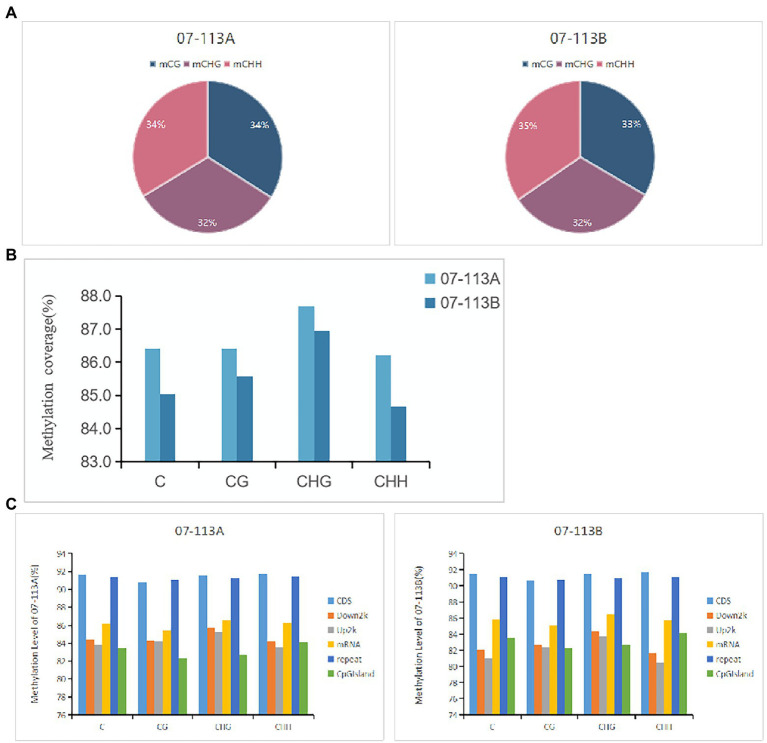
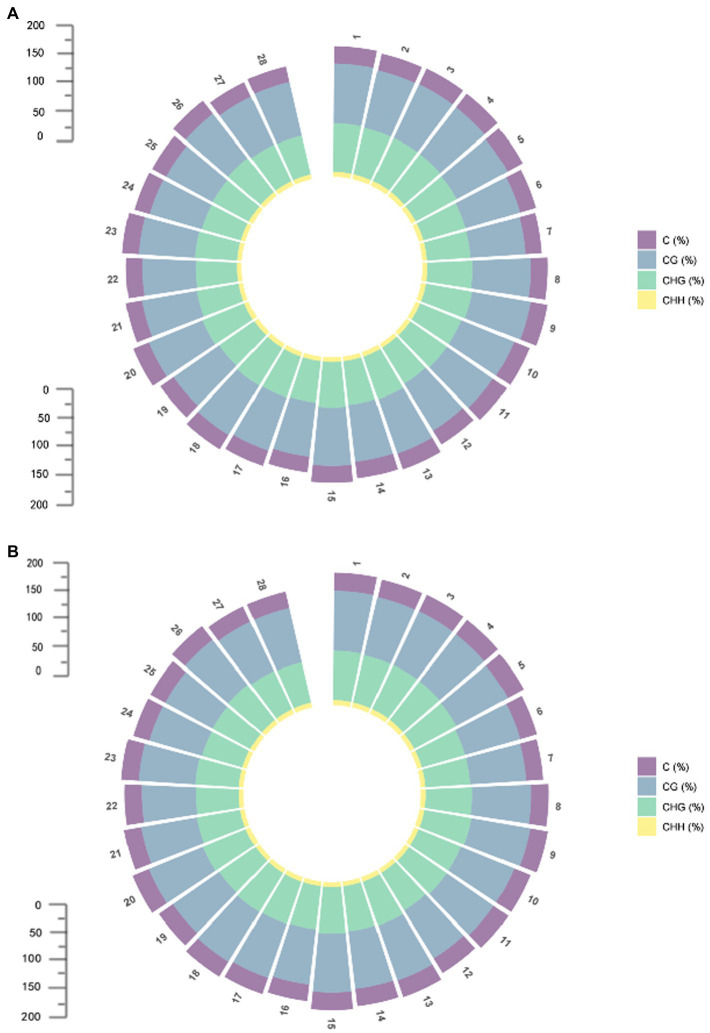
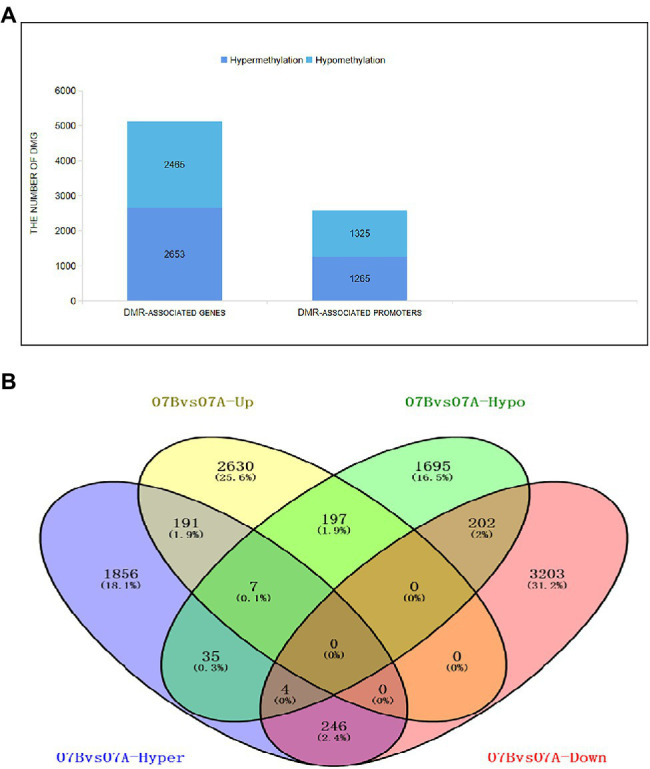
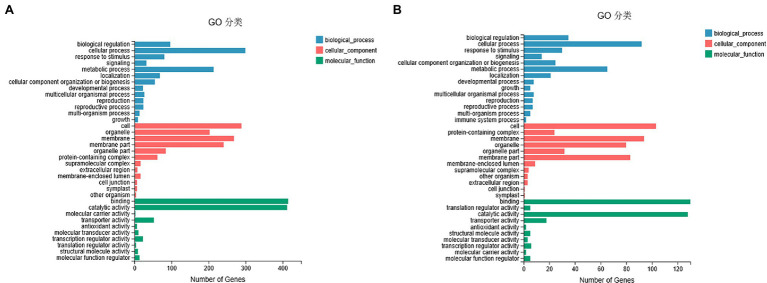
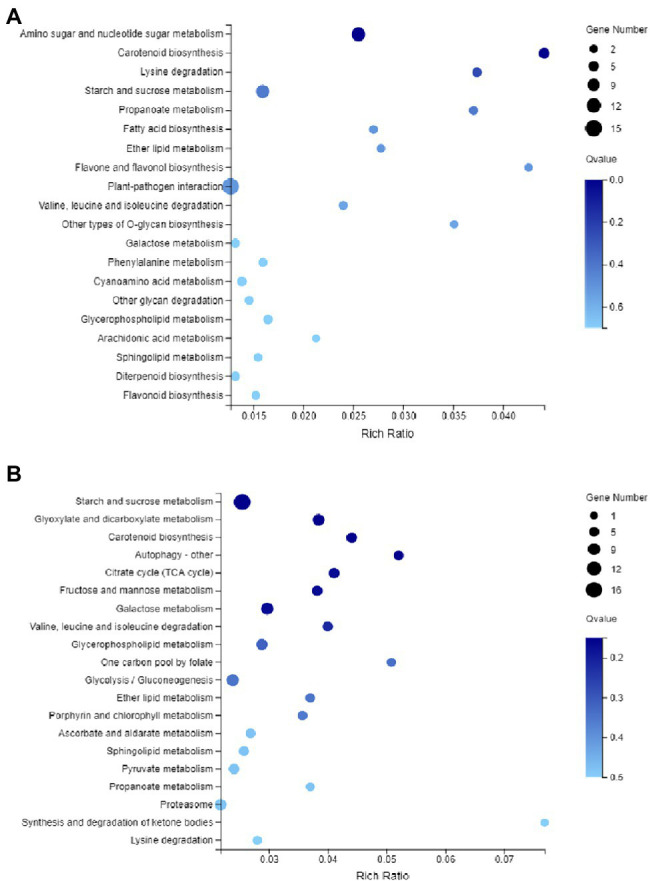
DNA methylation is defined as a conserved epigenetic modification mechanism that plays a key role in maintaining normal gene expression without altering the DNA sequence. Several studies have reported that altered methylation patterns were associated with male sterility in some plants such as rice and wheat, but global methylation profiles and their possible roles in cytoplasmic male sterility (CMS), especially in cotton near-isogenic lines, remain unclear. In this study, bisulfite sequencing technology and RNA-Seq were used to investigate CMS line 07-113A and its near-isogenic line 07-113B. Using integrated methylome and transcriptome analyses, we found that the number of hypermethylated genes in the differentially methylated regions, whether in the promoter region or in the gene region, was more in 07-113A than the number in 07-113B. The data indicated that 07-113A was more susceptible to methylation. In order to further analyze the regulatory network of male sterility, transcriptome sequencing and DNA methylation group data were used to compare the characteristics of near-isogenic lines 07-113A and 07-113B in cotton during the abortion stage. Combined methylation and transcriptome analysis showed that differentially expressed methylated genes were mainly concentrated in vital metabolic pathways including the starch and sucrose metabolism pathways and galactose metabolism. And there was a negative correlation between gene methylation and gene expression. In addition, five key genes that may be associated with CMS in cotton were identified. These data will support further understanding of the effect of DNA methylation on gene expression and their potential roles in cotton CMS.
DNA甲基化被定义为一种保守的表观遗传修饰机制,它在维持正常基因表达而不改变DNA序列方面起着关键作用。多项研究报告称,甲基化模式的改变与水稻和小麦等一些植物的雄性不育有关,但全基因组甲基化图谱及其在细胞质雄性不育(CMS)中的可能作用,尤其是在棉花近等基因系中的作用,仍不清楚。在本研究中,利用亚硫酸氢盐测序技术和RNA测序对CMS系07 - 113A及其近等基因系07 - 113B进行了研究。通过整合甲基化组和转录组分析,我们发现在差异甲基化区域(无论是启动子区域还是基因区域)中,07 - 113A的高甲基化基因数量比07 - 113B中的多。数据表明07 - 113A对甲基化更敏感。为了进一步分析雄性不育的调控网络,利用转录组测序和DNA甲基化组数据比较了棉花近等基因系07 - 113A和07 - 113B在败育阶段的特征。甲基化和转录组联合分析表明,差异表达的甲基化基因主要集中在包括淀粉和蔗糖代谢途径以及半乳糖代谢在内的重要代谢途径中。并且基因甲基化与基因表达之间存在负相关。此外,还鉴定了五个可能与棉花CMS相关的关键基因。这些数据将有助于进一步了解DNA甲基化对基因表达的影响及其在棉花CMS中的潜在作用。