Scientific Institute IRCCS E. MEDEA, Bioinformatics, Bosisio Parini, Italy.
Department of Biotechnology and Biosciences, University of Milan-Bicocca, Milan, Italy.
Mol Ecol. 2022 Jul;31(13):3672-3692. doi: 10.1111/mec.16531. Epub 2022 May 30.
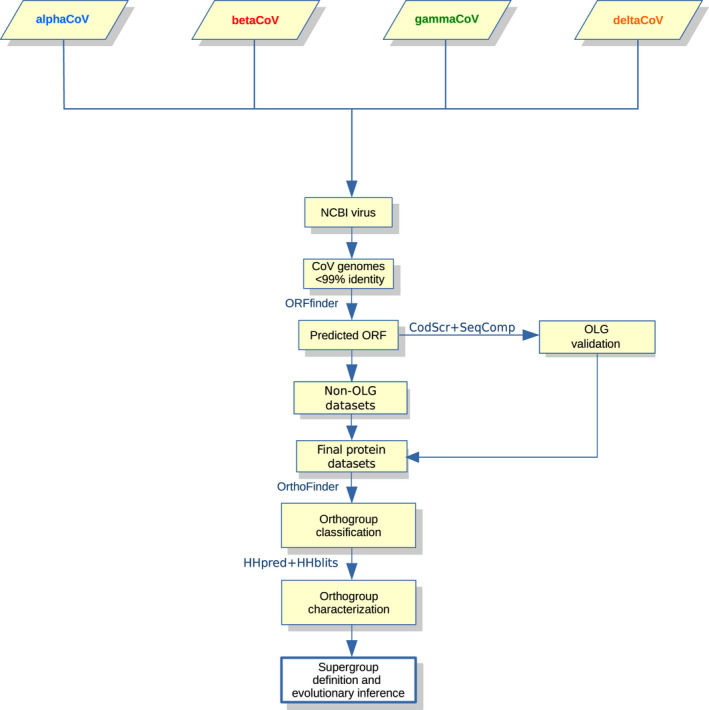
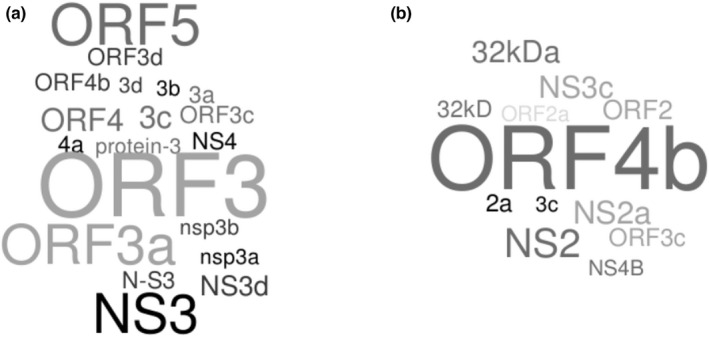
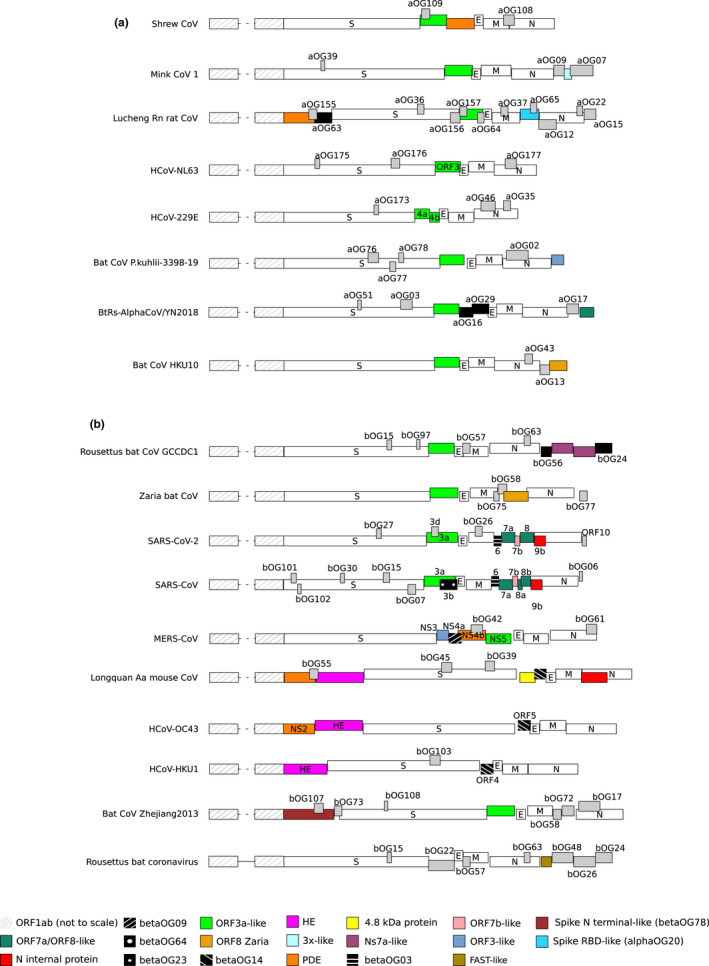
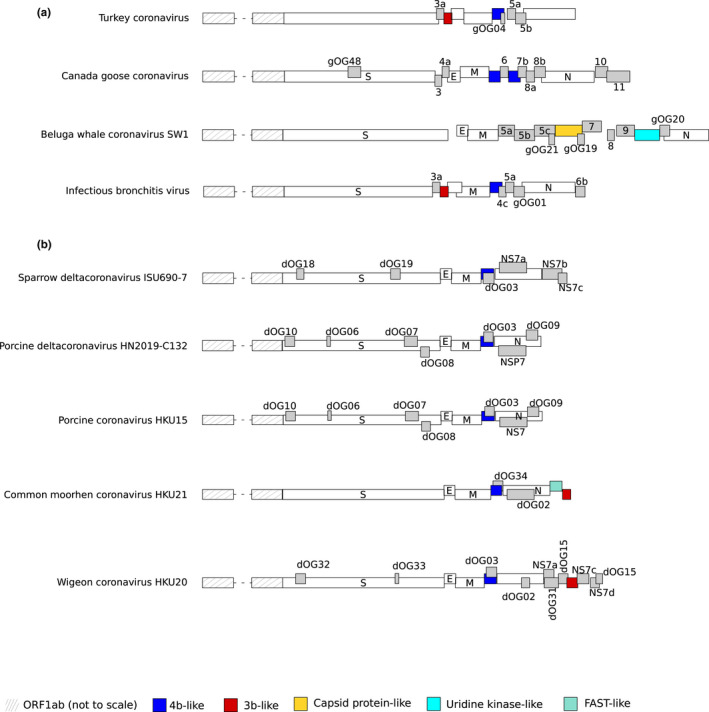
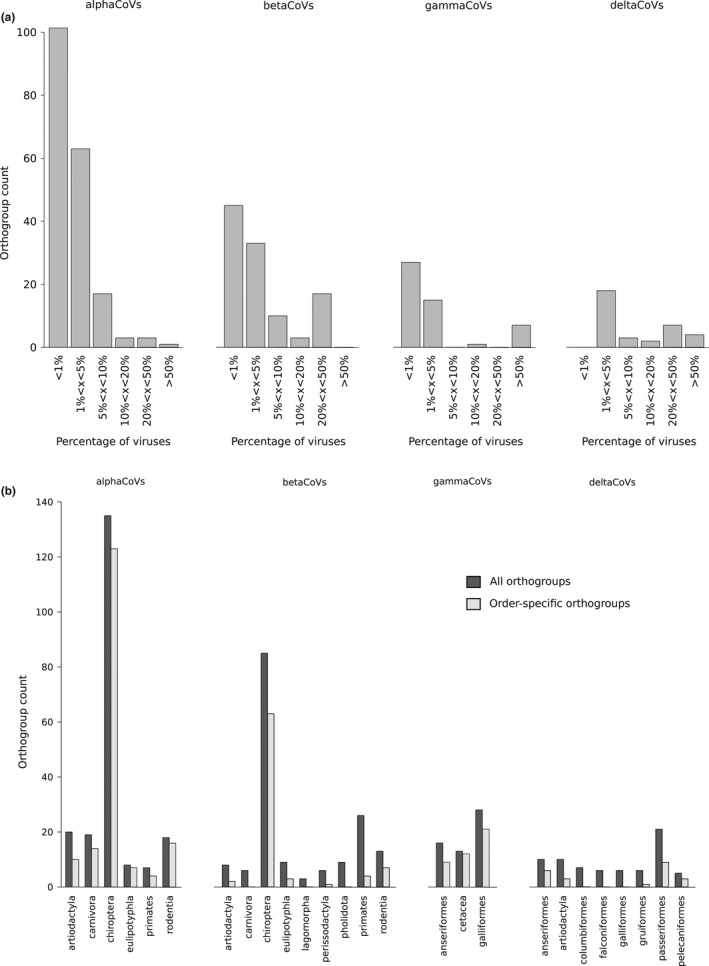
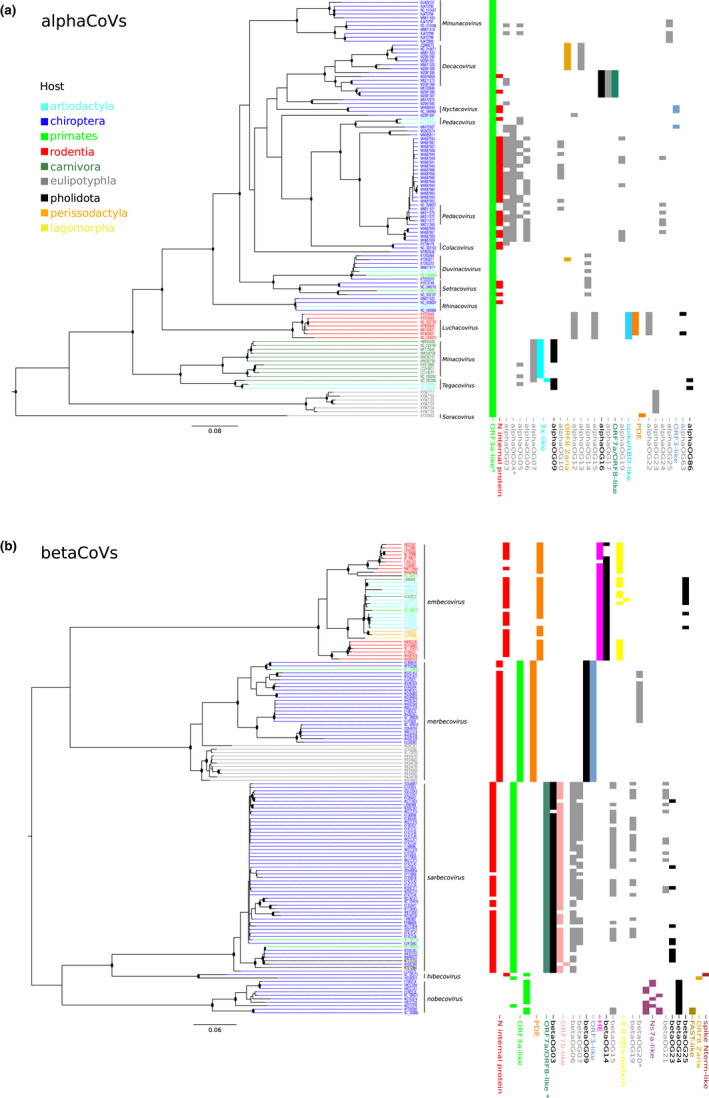
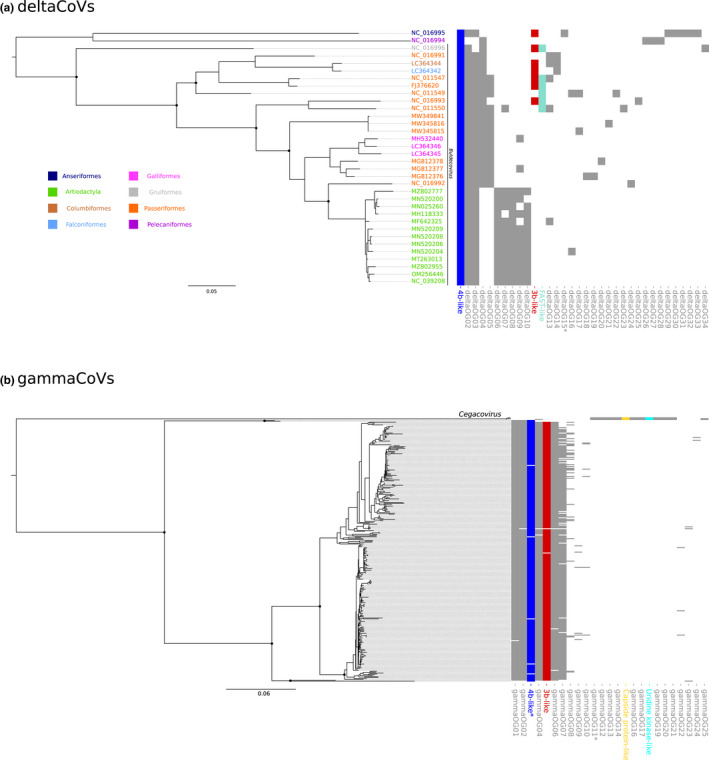
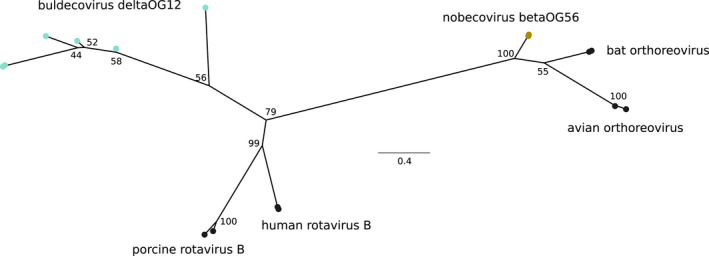
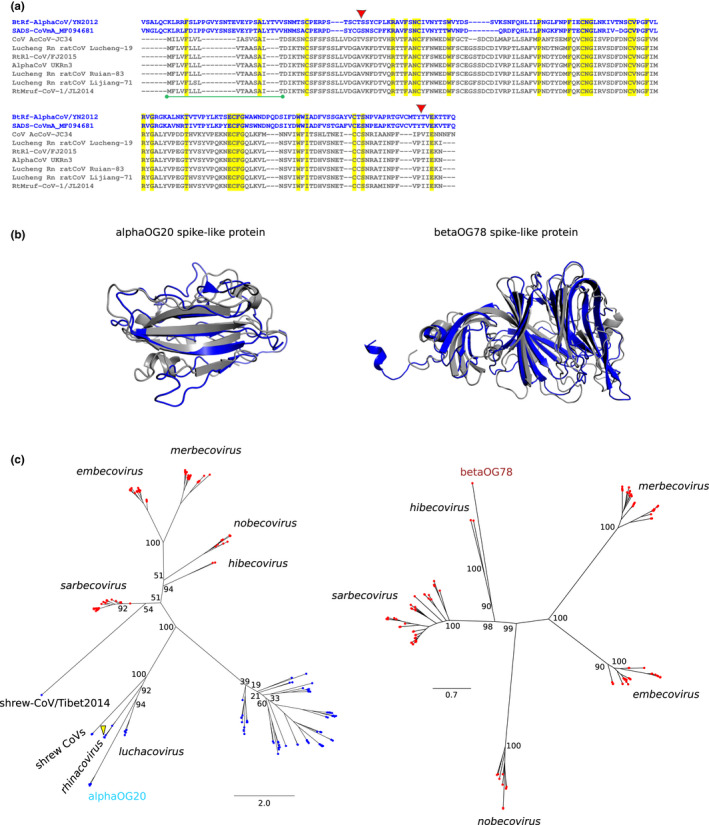
Coronaviruses (CoVs) have complex genomes that encode a fixed array of structural and nonstructural components, as well as a variety of accessory proteins that differ even among closely related viruses. Accessory proteins often play a role in the suppression of immune responses and may represent virulence factors. Despite their relevance for CoV phenotypic variability, information on accessory proteins is fragmentary. We applied a systematic approach based on homology detection to create a comprehensive catalogue of accessory proteins encoded by CoVs. Our analyses grouped accessory proteins into 379 orthogroups and 12 super-groups. No orthogroup was shared by the four CoV genera and very few were present in all or most viruses in the same genus, reflecting the dynamic evolution of CoV genomes. We observed differences in the distribution of accessory proteins in CoV genera. Alphacoronaviruses harboured the largest diversity of accessory open reading frames (ORFs), deltacoronaviruses the smallest. However, the average number of accessory proteins per genome was highest in betacoronaviruses. Analysis of the evolutionary history of some orthogroups indicated that the different CoV genera adopted similar evolutionary strategies. Thus, alphacoronaviruses and betacoronaviruses acquired phosphodiesterases and spike-like accessory proteins independently, whereas horizontal gene transfer from reoviruses endowed betacoronaviruses and deltacoronaviruses with fusion-associated small transmembrane (FAST) proteins. Finally, analysis of accessory ORFs in annotated CoV genomes indicated ambiguity in their naming. This complicates cross-communication among researchers and hinders automated searches of large data sets (e.g., PubMed, GenBank). We suggest that orthogroup membership is used together with a naming system to provide information on protein function.
冠状病毒(CoVs)具有复杂的基因组,可编码一系列固定的结构和非结构成分,以及多种不同的辅助蛋白,即使是密切相关的病毒之间也存在差异。辅助蛋白通常在抑制免疫反应方面发挥作用,可能是毒力因子。尽管它们与 CoV 表型变异性相关,但有关辅助蛋白的信息是零碎的。我们应用基于同源性检测的系统方法来创建 CoV 编码的辅助蛋白的综合目录。我们的分析将辅助蛋白分为 379 个直系同源物和 12 个超级组。四个 CoV 属之间没有共享的直系同源物,在同一属中,很少有存在于所有或大多数病毒中的直系同源物,反映了 CoV 基因组的动态进化。我们观察到 CoV 属中辅助蛋白的分布存在差异。α冠状病毒具有最大多样性的辅助开放阅读框(ORF),δ冠状病毒具有最小多样性。然而,每个基因组的辅助蛋白数量平均值在β冠状病毒中最高。对一些直系同源物的进化历史进行分析表明,不同的 CoV 属采用了相似的进化策略。因此,α冠状病毒和β冠状病毒独立地获得了磷酸二酯酶和类似刺突的辅助蛋白,而来自呼肠孤病毒的水平基因转移使β冠状病毒和δ冠状病毒获得了融合相关的小跨膜(FAST)蛋白。最后,对注释 CoV 基因组中的辅助 ORF 进行分析表明,它们的命名存在歧义。这使得研究人员之间的交流变得复杂,并阻碍了对大型数据集(例如 PubMed、GenBank)的自动搜索。我们建议使用直系同源物成员资格以及命名系统一起提供有关蛋白质功能的信息。