Department of Gynecology, Cancer Hospital of China Medical University, Liaoning Cancer Hospital & Institute, No. 44 Xiaoheyan Road, Dadong District, Shenyang, 110042 Liaoning Province, China.
Comput Math Methods Med. 2022 May 5;2022:8338137. doi: 10.1155/2022/8338137. eCollection 2022.
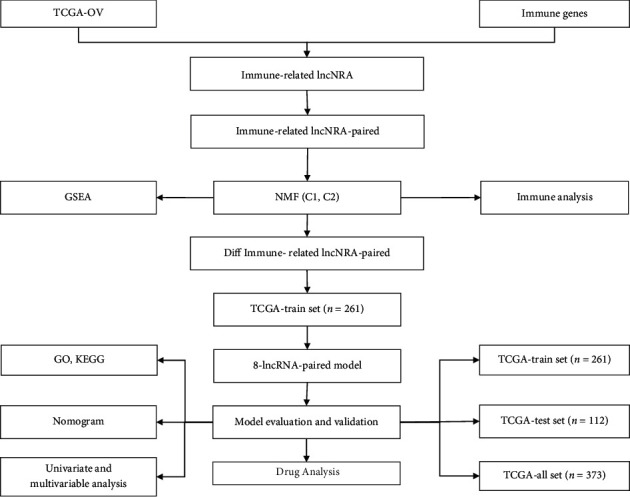
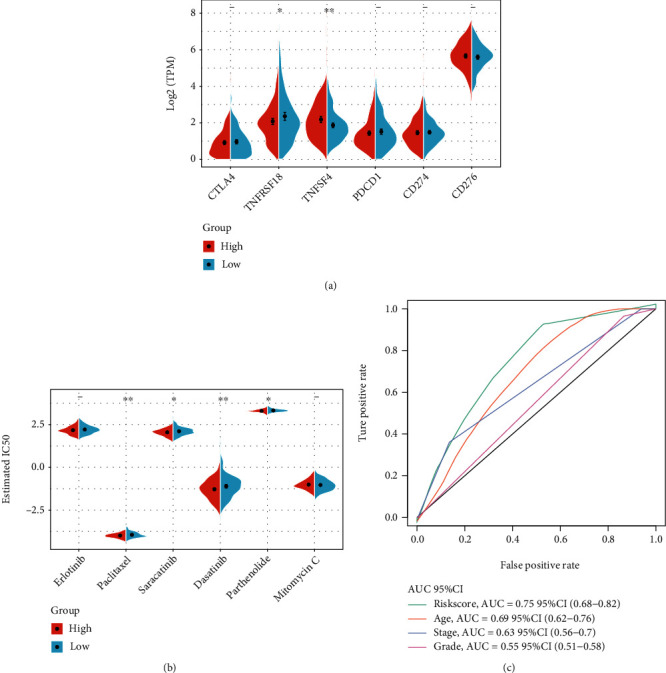
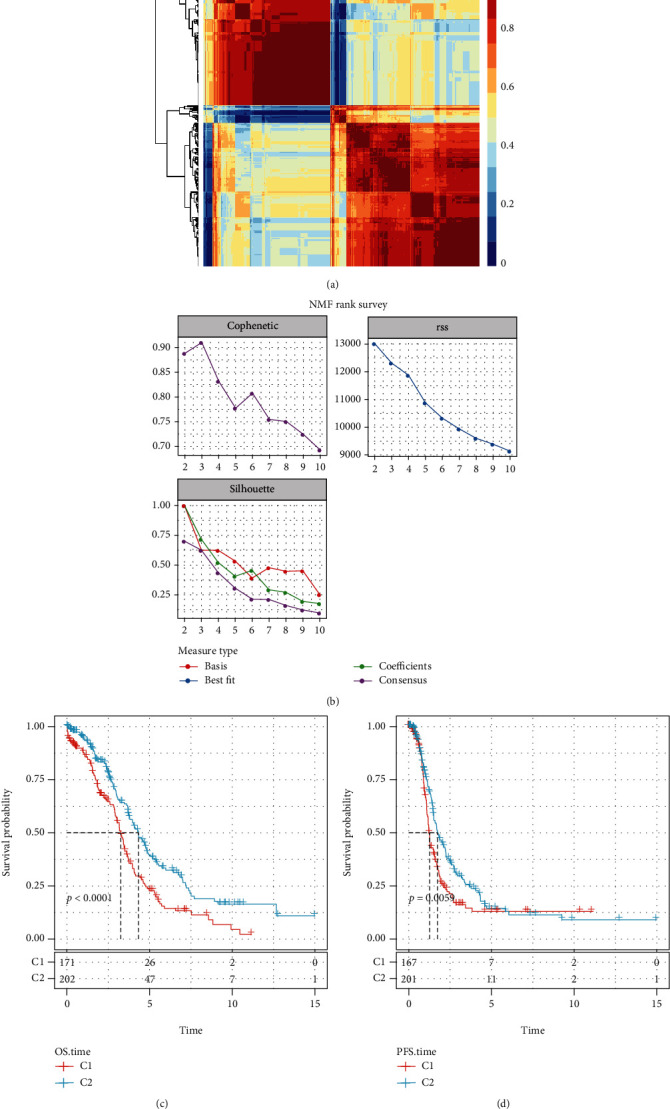
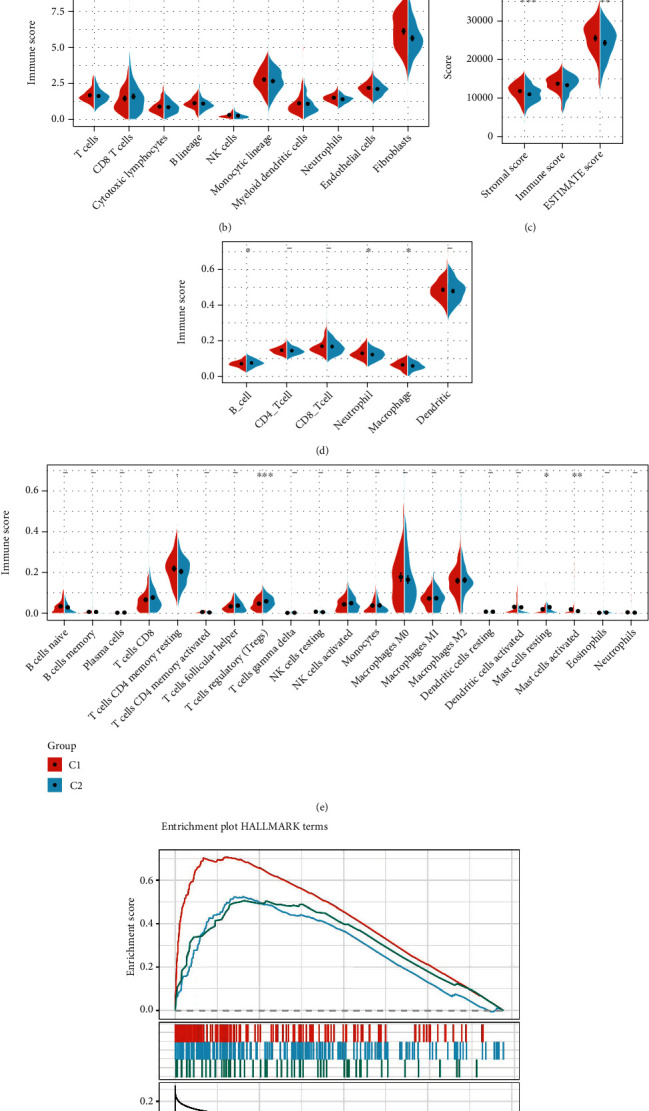
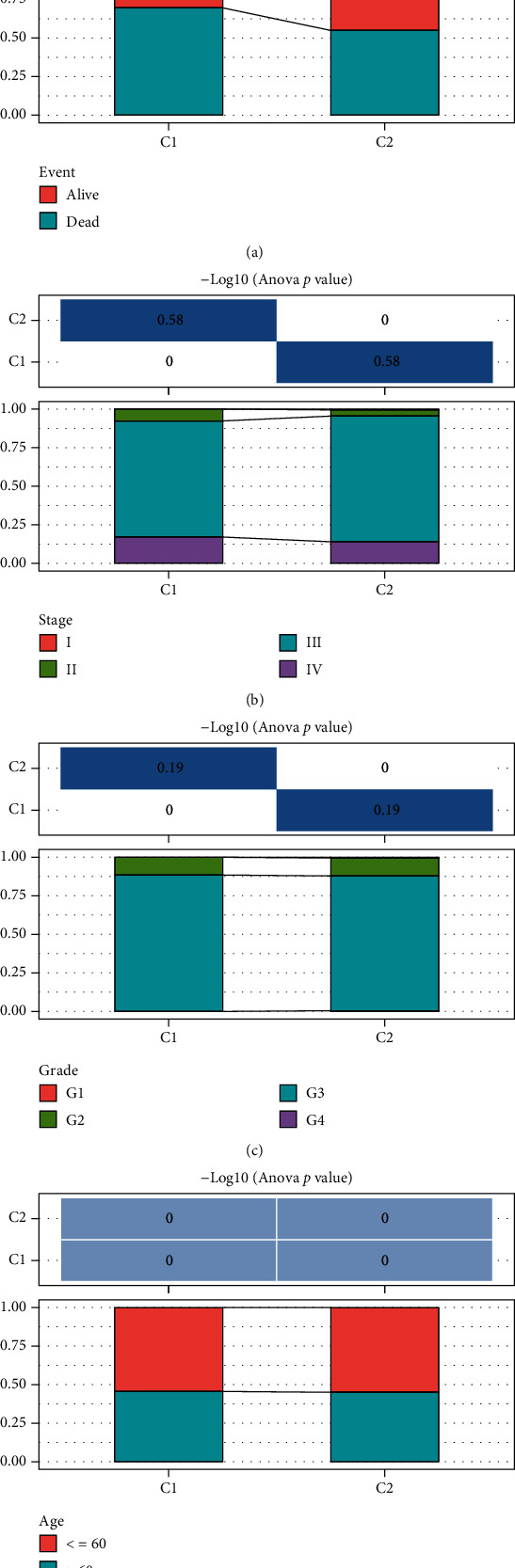
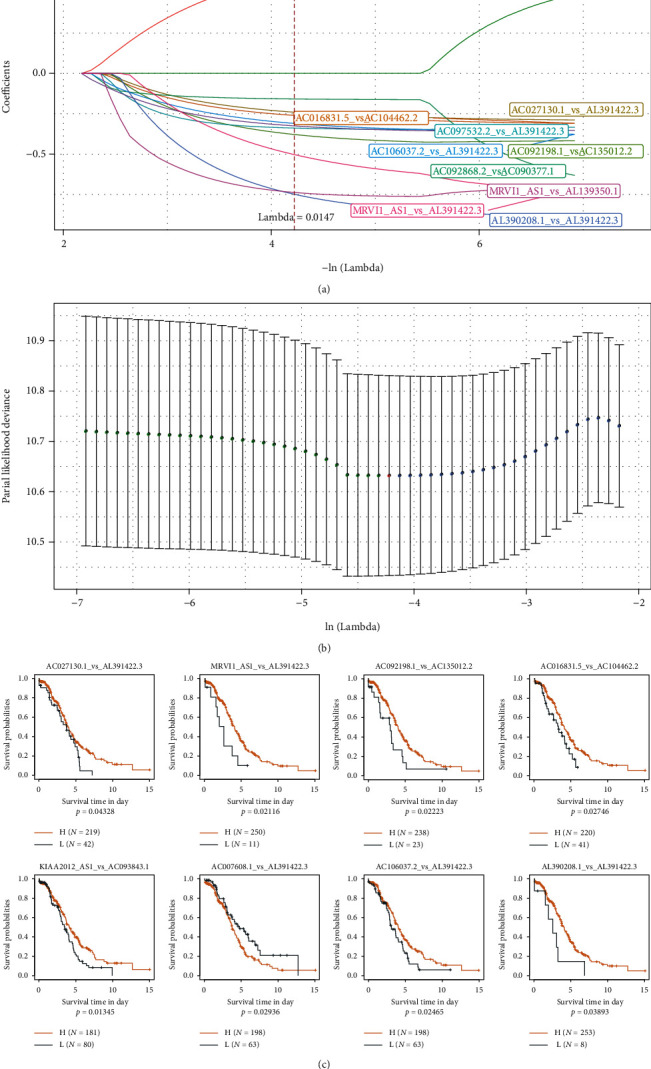
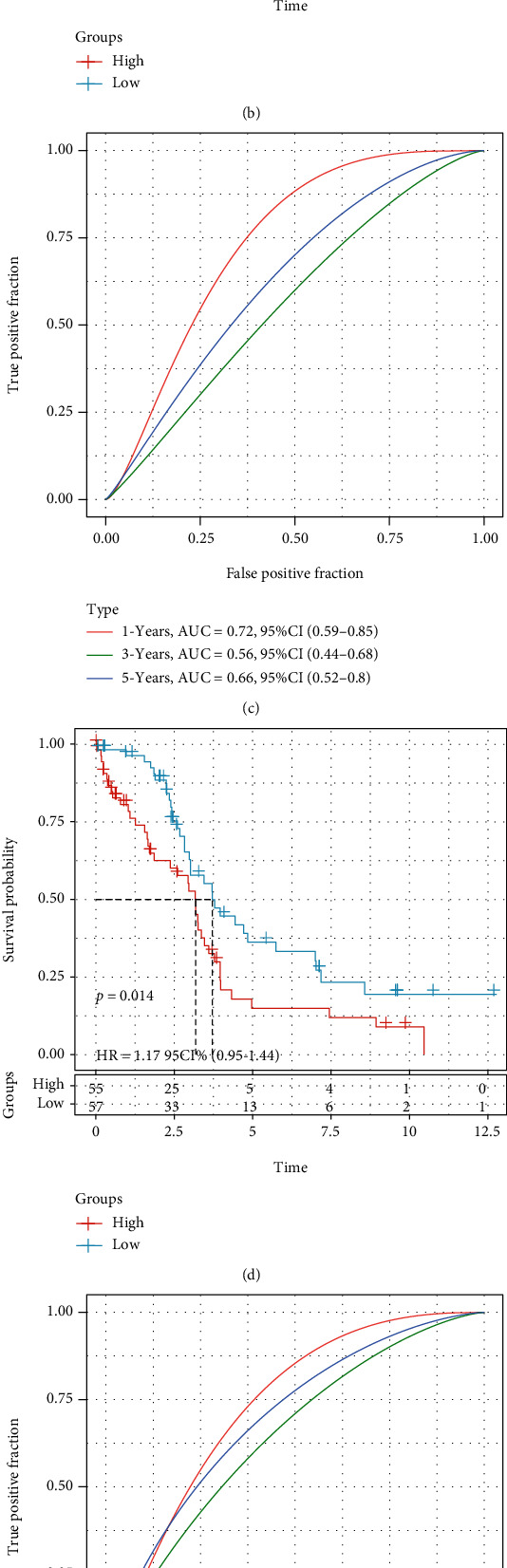
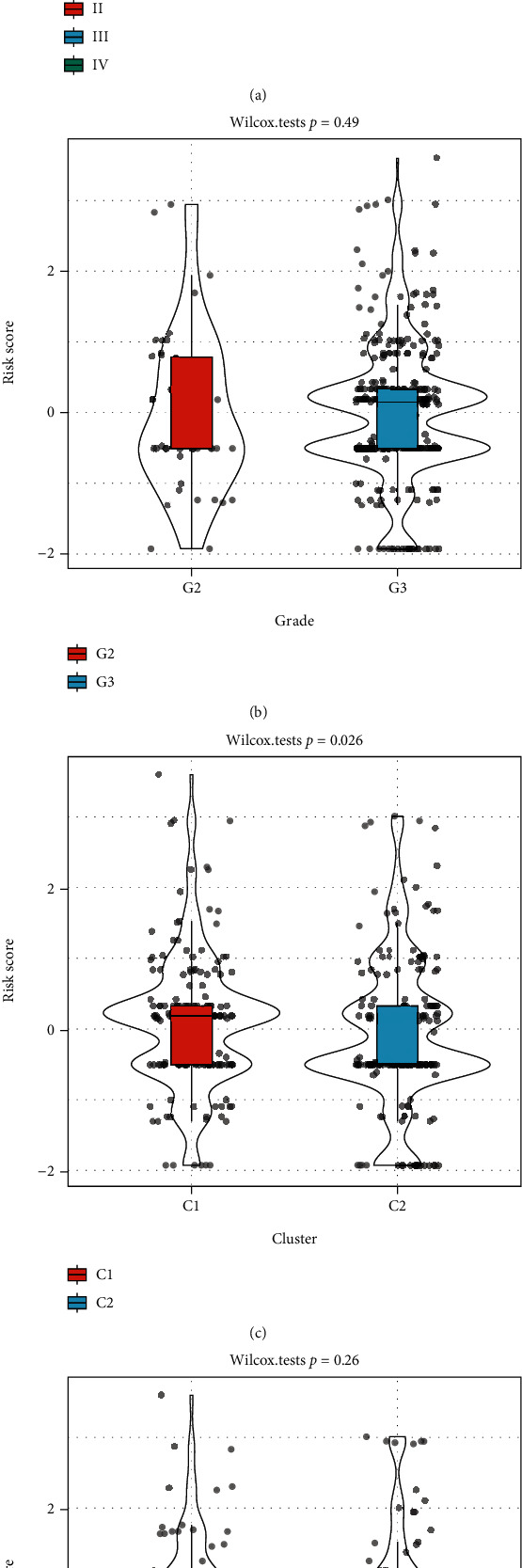
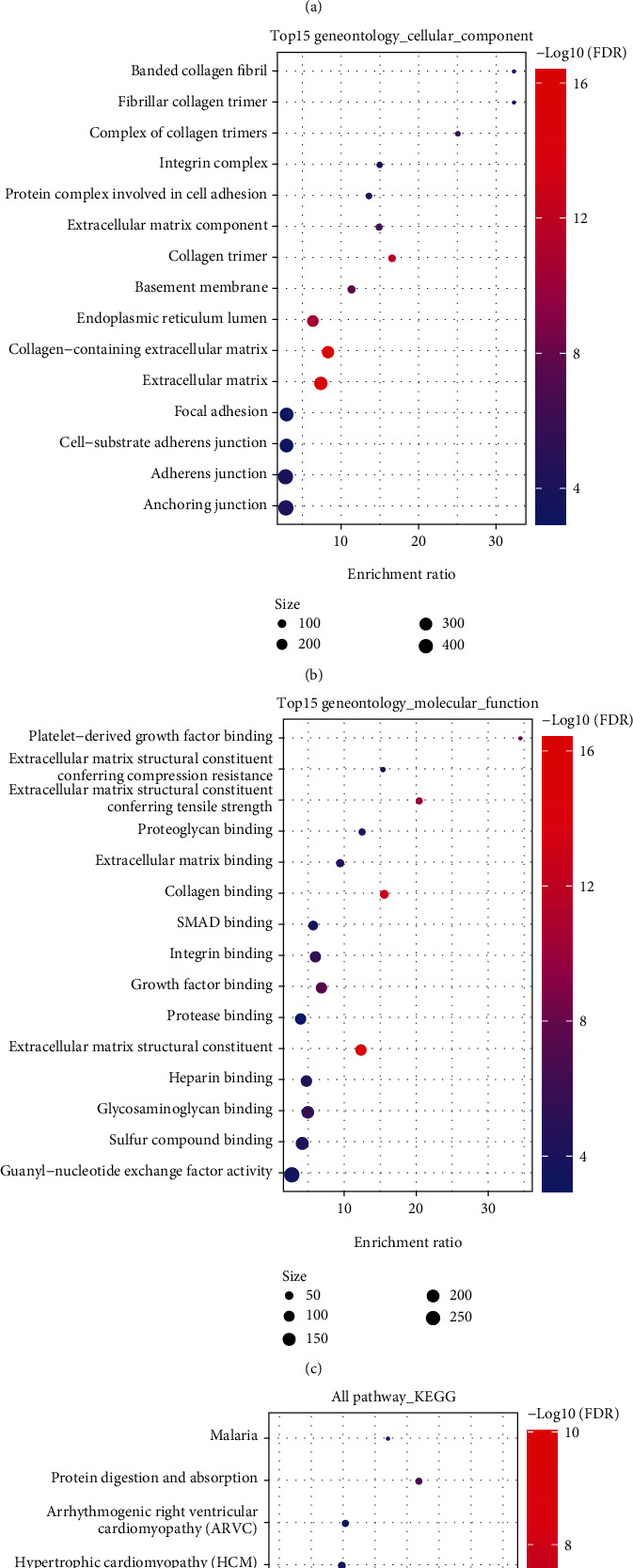
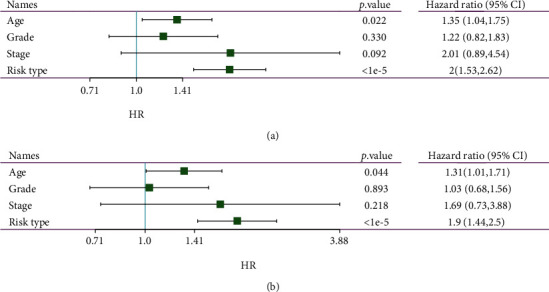
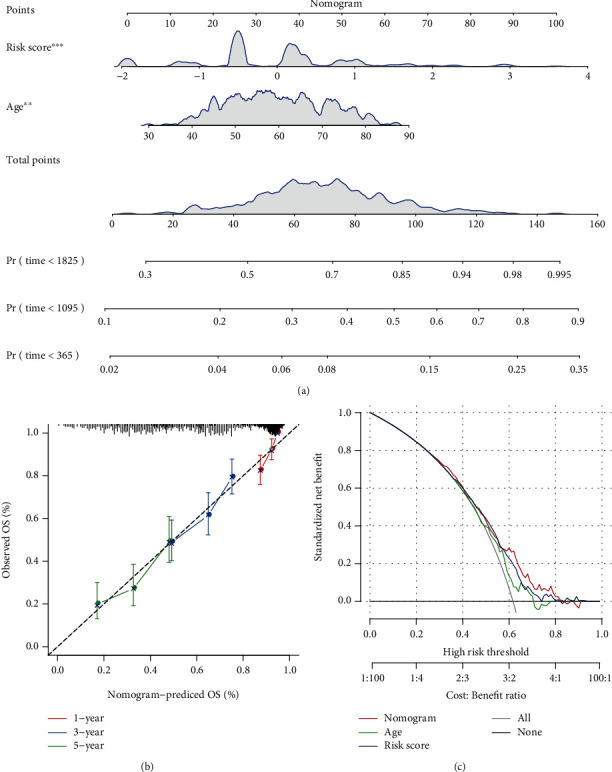
This study collected immune-related genes (IRGs) and used gene expression data from TCGA database to construct a molecular subtype of ovarian cancer (OV) based on immune-related lncRNA gene pairs (IRLnc_GPs). The relationships between molecular subtypes and prognosis and clinical characteristics were further explored. IRGs were acquired from the ImmPort database, and round-robin pairing of immune-related lncRNAs was performed. The NMF algorithm was used to identify molecular subtypes, and the immune score of a single sample was calculated through ESTIMATE, TIMER, ssGSEA, MCPcounter, and CIBERSORT. The relationship between molecular subtypes and immune microenvironments was identified. A hypergeometric test was used to test the lncRNA pairs among the OV molecular subtypes (C1 and C2 subtypes). The BH method was used to screen the different lncRNA pairs, and a predictive risk model was constructed and verified. Finally, correlation analysis between the risk model, immune checkpoint genes, and chemotherapy drugs was carried out. Based on IRLnc_GP to classify 373 OV samples of TCGA, the samples were divided into two subtypes, and the prognosis between the subtypes showed significant differences. The C1 subtype with a poor prognosis was more related to the pathways of tumor occurrence and development. We identified 180 differential lncRNA pairs between subtypes and constructed a prognostic risk model based on 8 IRLnc_GPs. In the independent dataset, the distribution of subtypes in functional modules was different and highly repeatable. There were significant differences in the molecular and clinical characteristics of the subtypes and the drug sensitivity of immunotherapy/chemotherapy. In conclusion, the risk model established based on IRLnc_GP can better evaluate the prognosis of OV samples and can also assess the effects of different drug treatments in the high- and low-risk groups, providing new insights and ideas for the treatment of OV.
本研究收集免疫相关基因(IRGs),并利用 TCGA 数据库中的基因表达数据,基于免疫相关长链非编码 RNA 基因对(IRLnc_GPs)构建卵巢癌(OV)的分子亚型。进一步探讨了分子亚型与预后和临床特征的关系。IRGs 从 ImmPort 数据库中获取,对免疫相关长链非编码 RNA 进行轮询配对。采用 NMF 算法识别分子亚型,通过 ESTIMATE、TIMER、ssGSEA、MCPcounter 和 CIBERSORT 计算单个样本的免疫评分。鉴定分子亚型与免疫微环境的关系。使用超几何检验测试 OV 分子亚型(C1 和 C2 亚型)之间的长链非编码 RNA 对。使用 BH 方法筛选不同的长链非编码 RNA 对,构建并验证预测风险模型。最后,对风险模型、免疫检查点基因和化疗药物进行相关性分析。基于 IRLnc_GP 对 TCGA 的 373 个 OV 样本进行分类,将样本分为两种亚型,且两种亚型的预后差异具有统计学意义。预后较差的 C1 亚型与肿瘤发生发展的途径更为相关。我们鉴定了 180 对亚型间差异的长链非编码 RNA 对,并基于 8 个 IRLnc_GPs 构建了预后风险模型。在独立数据集上,功能模块中亚型的分布不同且高度可重复。亚型之间的分子和临床特征以及免疫治疗/化疗的药物敏感性存在显著差异。总之,基于 IRLnc_GP 建立的风险模型可以更好地评估 OV 样本的预后,还可以评估高、低风险组中不同药物治疗的效果,为 OV 的治疗提供新的思路和见解。