Lewis-Sigler Institute for Integrative Genomics, Carl Icahn Laboratory, Princeton University, South Drive, Princeton, NJ 08540, USA.
Department of Molecular & Cellular Biology, Harvard University, Divinity Avenue, Cambridge, MA 02138, USA.
Curr Biol. 2022 Jun 20;32(12):2632-2639.e2. doi: 10.1016/j.cub.2022.04.085. Epub 2022 May 18.
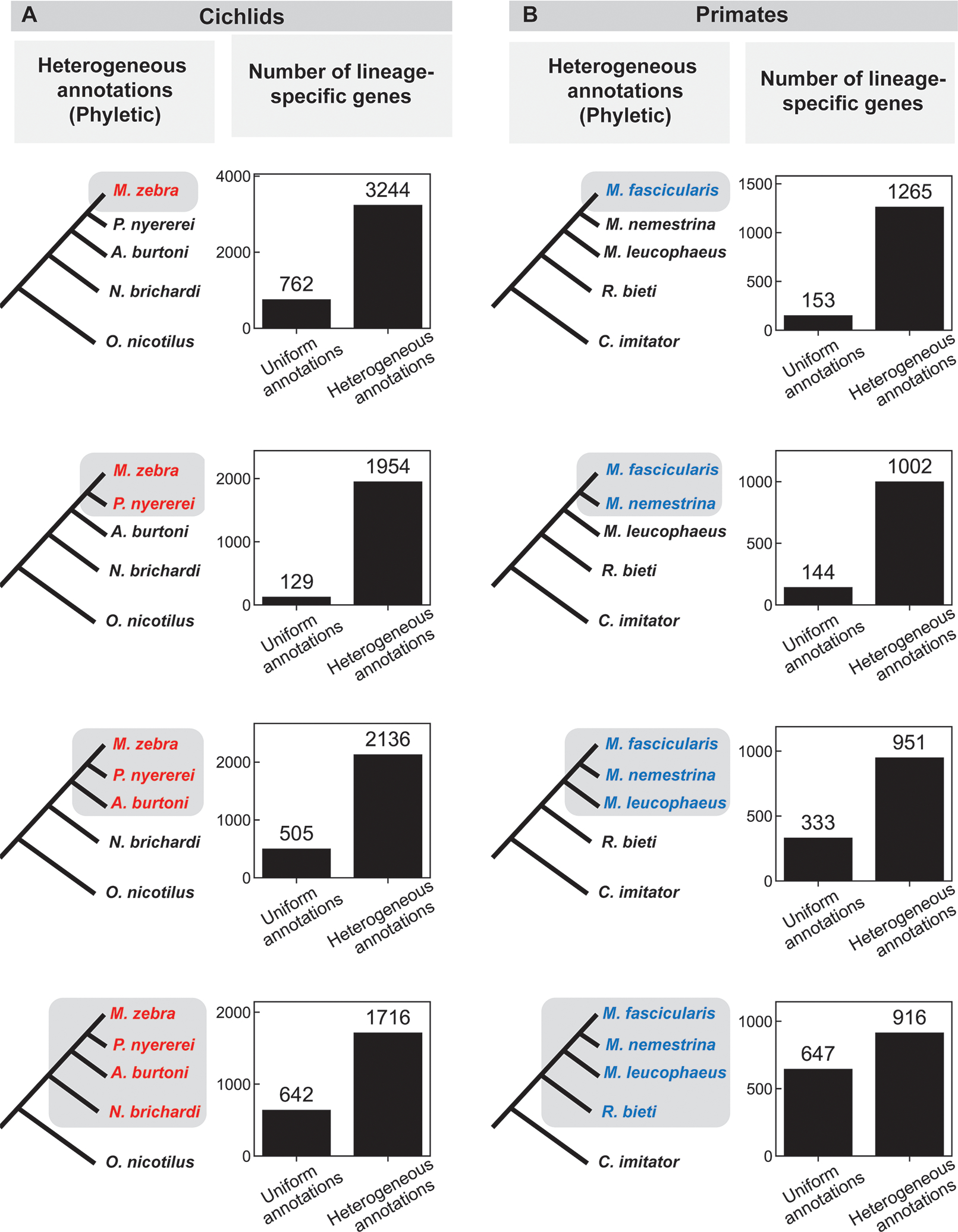
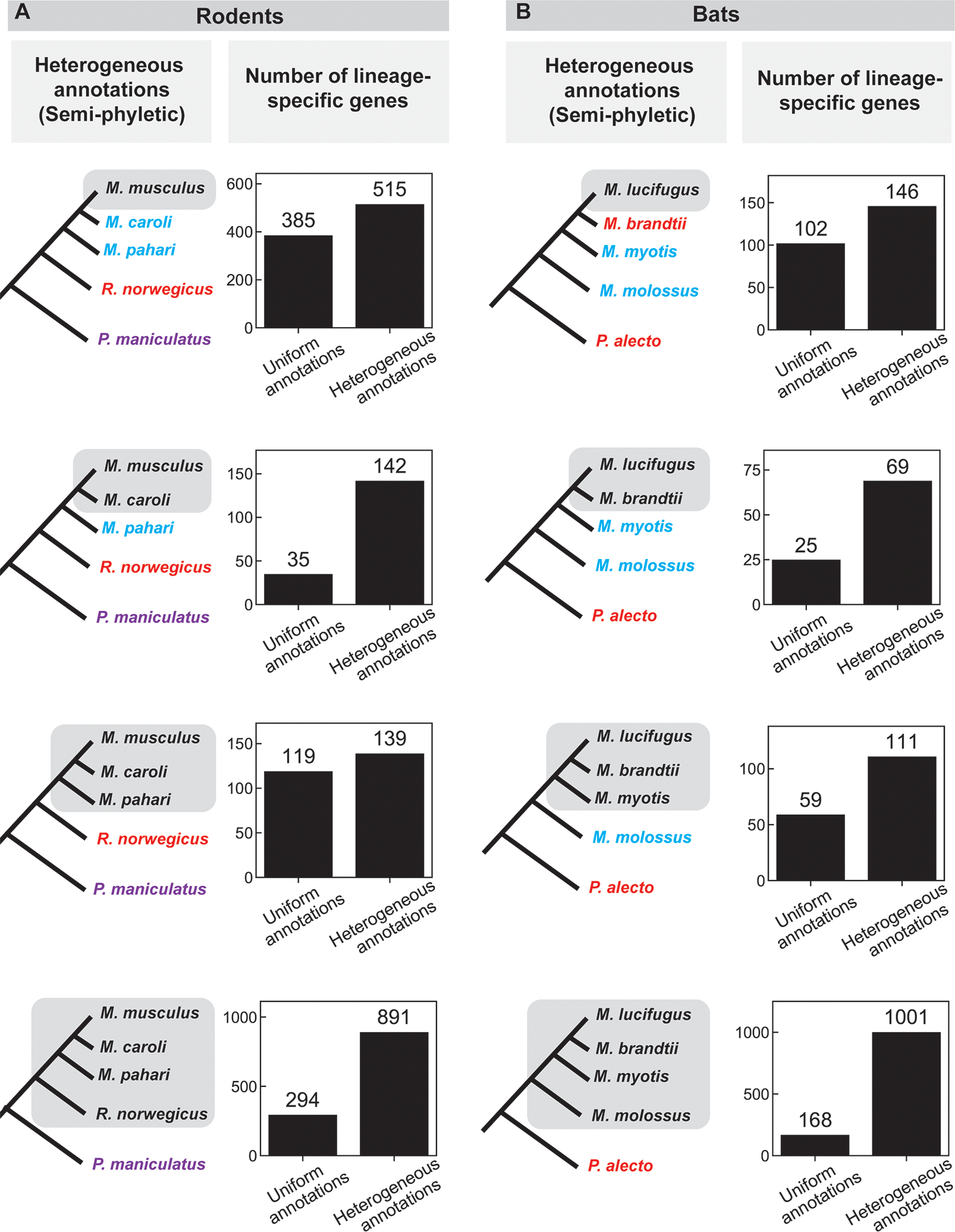
Comparisons of genomes of different species are used to identify lineage-specific genes, those genes that appear unique to one species or clade. Lineage-specific genes are often thought to represent genetic novelty that underlies unique adaptations. Identification of these genes depends not only on genome sequences, but also on inferred gene annotations. Comparative analyses typically use available genomes that have been annotated using different methods, increasing the risk that orthologous DNA sequences may be erroneously annotated as a gene in one species but not another, appearing lineage specific as a result. To evaluate the impact of such "annotation heterogeneity," we identified four clades of species with sequenced genomes with more than one publicly available gene annotation, allowing us to compare the number of lineage-specific genes inferred when differing annotation methods are used to those resulting when annotation method is uniform across the clade. In these case studies, annotation heterogeneity increases the apparent number of lineage-specific genes by up to 15-fold, suggesting that annotation heterogeneity is a substantial source of potential artifact.
对不同物种基因组的比较用于鉴定谱系特异性基因,即那些出现在一个物种或进化枝中独特的基因。通常认为,谱系特异性基因代表了遗传新颖性,是独特适应性的基础。这些基因的鉴定不仅取决于基因组序列,还取决于推断的基因注释。比较分析通常使用已注释的可用基因组,但使用的方法不同,增加了以下风险:同源 DNA 序列可能在一个物种中错误地注释为一个基因,但在另一个物种中却没有,从而表现出谱系特异性。为了评估这种“注释异质性”的影响,我们确定了四个具有多个公开可用基因注释的测序物种进化枝,这使我们能够比较在使用不同注释方法时推断出的谱系特异性基因的数量与在整个进化枝中使用统一注释方法时的数量。在这些案例研究中,注释异质性使谱系特异性基因的数量最多增加了 15 倍,这表明注释异质性是潜在人工制品的一个重要来源。