Nora Eccles Harrison Cardiovascular Research and Training Institute, University of Utah, Salt Lake City, UT, USA.
Molecular Medicine Program, University of Utah, Salt Lake City, UT, USA.
Nat Commun. 2022 May 19;13(1):2769. doi: 10.1038/s41467-022-30236-4.
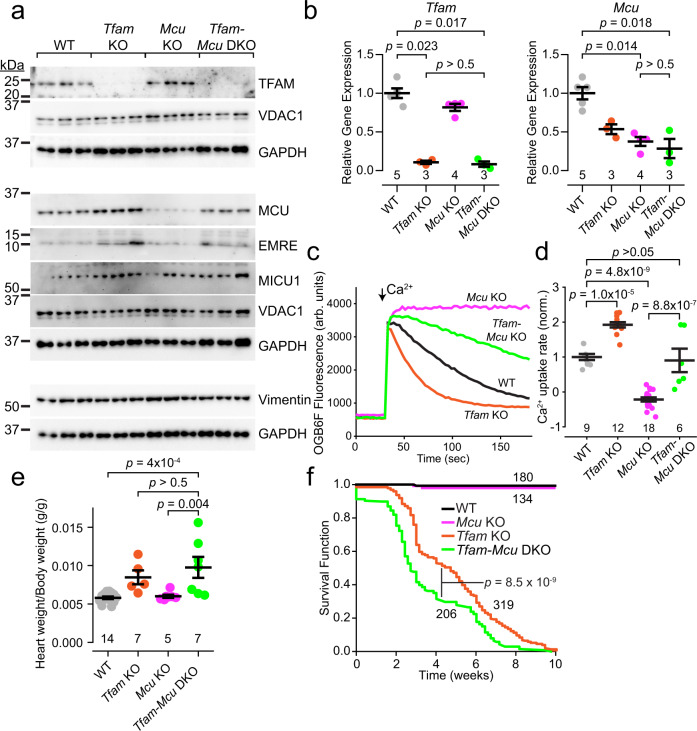
Calcium entering mitochondria potently stimulates ATP synthesis. Increases in calcium preserve energy synthesis in cardiomyopathies caused by mitochondrial dysfunction, and occur due to enhanced activity of the mitochondrial calcium uniporter channel. The signaling mechanism that mediates this compensatory increase remains unknown. Here, we find that increases in the uniporter are due to impairment in Complex I of the electron transport chain. In normal physiology, Complex I promotes uniporter degradation via an interaction with the uniporter pore-forming subunit, a process we term Complex I-induced protein turnover. When Complex I dysfunction ensues, contact with the uniporter is inhibited, preventing degradation, and leading to a build-up in functional channels. Preventing uniporter activity leads to early demise in Complex I-deficient animals. Conversely, enhancing uniporter stability rescues survival and function in Complex I deficiency. Taken together, our data identify a fundamental pathway producing compensatory increases in calcium influx during Complex I impairment.
钙进入线粒体可强力刺激 ATP 合成。钙的增加可在由线粒体功能障碍引起的心肌病中保存能量合成,并且由于线粒体钙单向转运体通道的活性增强而发生。介导这种代偿性增加的信号机制尚不清楚。在这里,我们发现单向转运体的增加是由于电子传递链复合物 I 的损伤所致。在正常生理学中,复合物 I 通过与单向转运体孔形成亚基的相互作用促进单向转运体的降解,这一过程我们称之为复合物 I 诱导的蛋白周转。当复合物 I 功能障碍发生时,与单向转运体的接触受到抑制,防止降解,并导致功能性通道的积累。阻止单向转运体的活性会导致复合物 I 缺陷动物的早期死亡。相反,增强单向转运体的稳定性可挽救复合物 I 缺乏症中的存活和功能。总之,我们的数据确定了一条基本途径,即在复合物 I 损伤期间产生钙流入的代偿性增加。