Department of Science and Technology, Federal University of São Paulo, São José dos Campos, Brazil.
Laboratory of Applied Toxinology, Center of Toxins, Immune-Response and Cell Signaling (CeTICS), Butantan Institute, São Paulo, Brazil.
Peptides. 2022 Aug;154:170814. doi: 10.1016/j.peptides.2022.170814. Epub 2022 May 26.
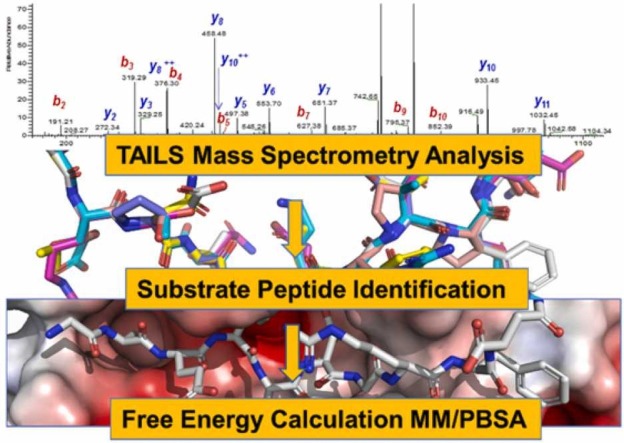
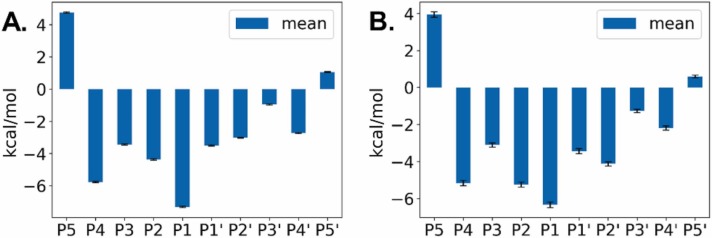
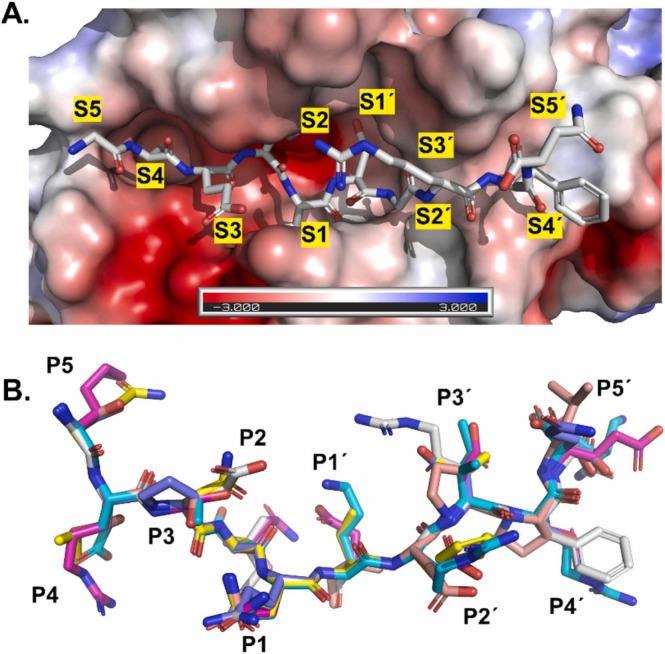
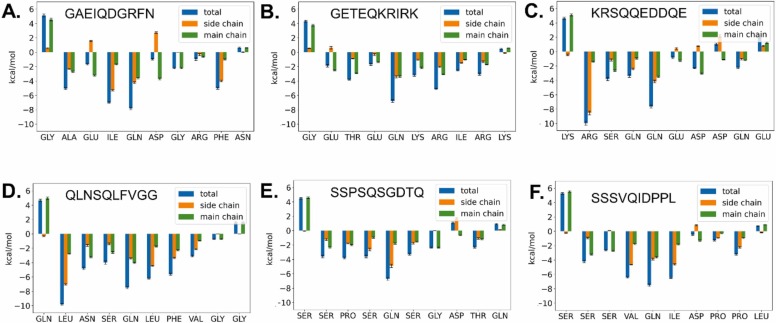
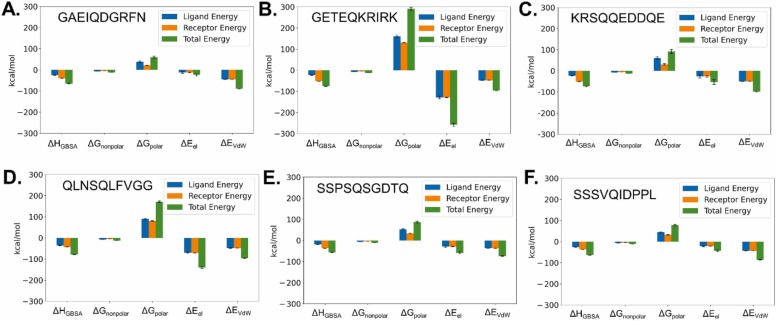
The main protease M of SARS-CoV-2 is a well-studied major drug target. Additionally, it has been linked to this virus' pathogenicity, possibly through off-target effects. It is also an interesting diagnostic target. To obtain more data on possible substrates as well as to assess the enzyme's primary specificity a two-step approach was introduced. First, Terminal Amine Isobaric Labeling of Substrates (TAILS) was employed to identify novel M cleavage sites in a mouse lung proteome library. In a second step, using a structural homology model, the MM/PBSA variant MM/GBSA (Molecular Mechanics Poisson-Boltzmann/Generalized Born Surface Area) free binding energy calculations were carried out to determine relevant interacting amino acids. As a result, 58 unique cleavage sites were detected, including six that displayed glutamine at the P1 position. Furthermore, modeling results indicated that M has a far higher potential promiscuity towards substrates than expected. The combination of proteomics and MM/PBSA modeling analysis can thus be useful for elucidating the specificity of M, and thus open novel perspectives for the development of future peptidomimetic drugs against COVID-19, as well as diagnostic tools.
新型冠状病毒 2 的主要蛋白酶 M 是一个研究得很好的主要药物靶点。此外,它与该病毒的致病性有关,可能通过非靶效应。它也是一个有趣的诊断靶点。为了获得更多关于可能的底物的数据,并评估酶的主要特异性,采用了两步法。首先,采用末端胺同重标记的底物(TAILS)方法,在小鼠肺蛋白组文库中鉴定新型 M 切割位点。在第二步中,使用结构同源模型,进行了分子力学泊松-玻尔兹曼/广义 Born 表面面积(MM/PBSA)自由结合能计算,以确定相关的相互作用氨基酸。结果,检测到 58 个独特的切割位点,包括 6 个在 P1 位置显示谷氨酰胺的位点。此外,建模结果表明,M 对底物的潜在混杂性远远高于预期。因此,蛋白质组学和 MM/PBSA 建模分析的结合可用于阐明 M 的特异性,从而为开发针对 COVID-19 的未来肽模拟药物以及诊断工具开辟新的前景。