Division of Biosciences, University College London, London WC1E 6BT, United Kingdom.
Institute of Structural and Molecular Biology, Birkbeck College, London WC1E 7HX, United Kingdom.
ACS Chem Neurosci. 2022 Jun 15;13(12):1805-1817. doi: 10.1021/acschemneuro.2c00200. Epub 2022 Jun 3.
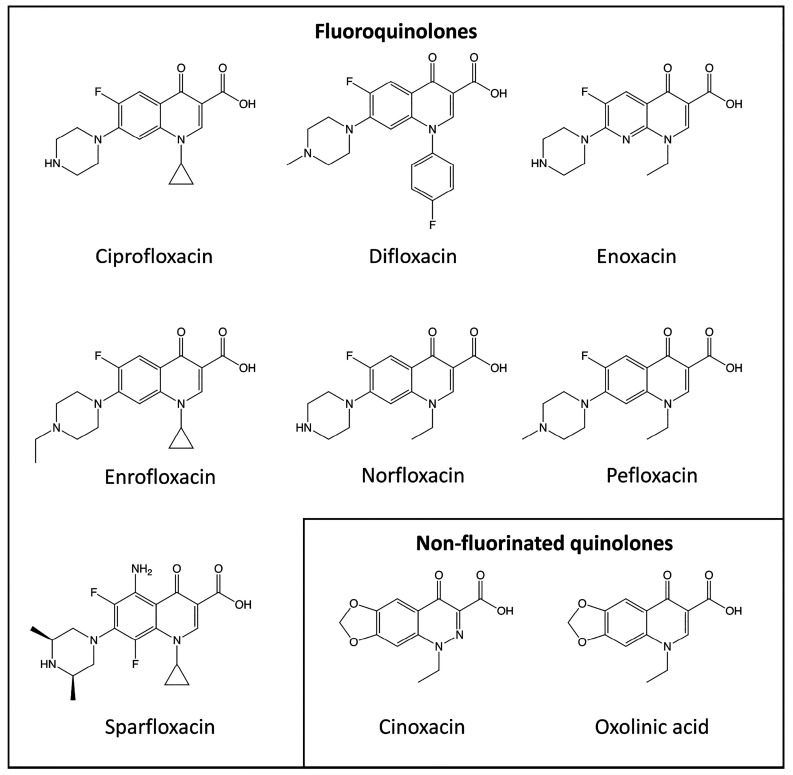
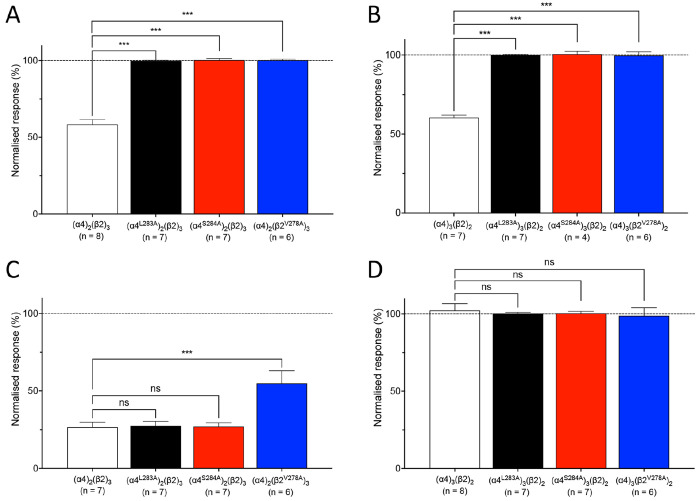
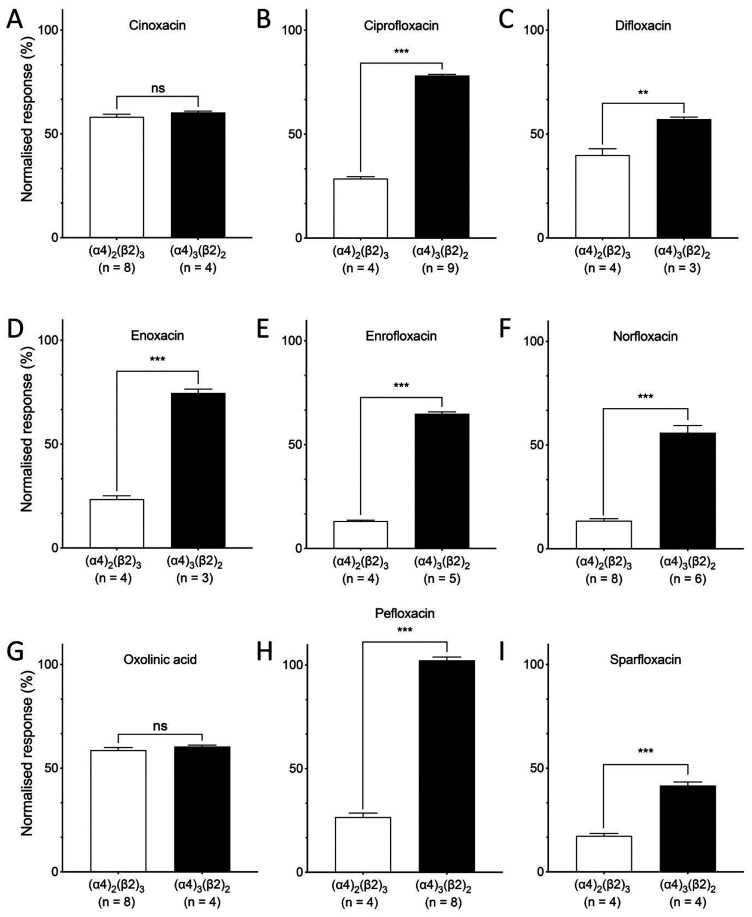
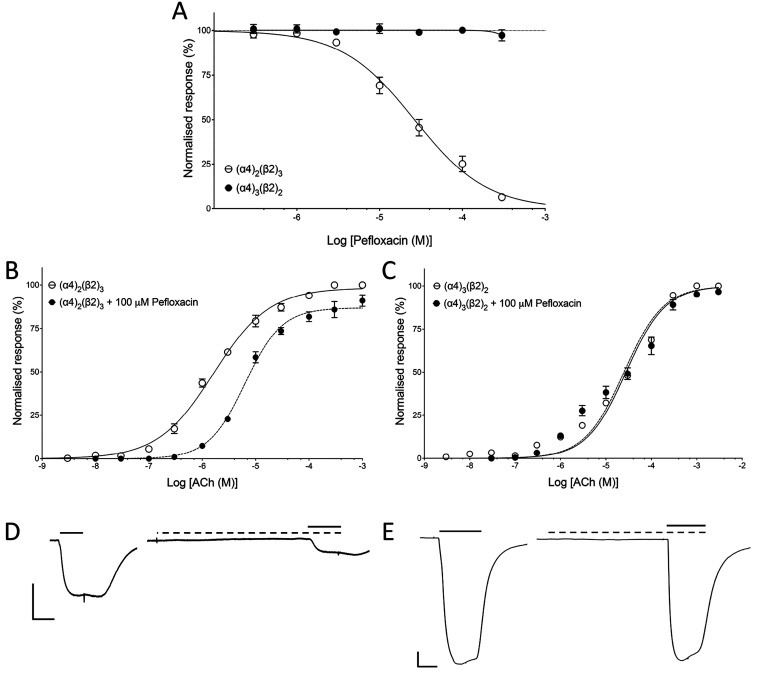
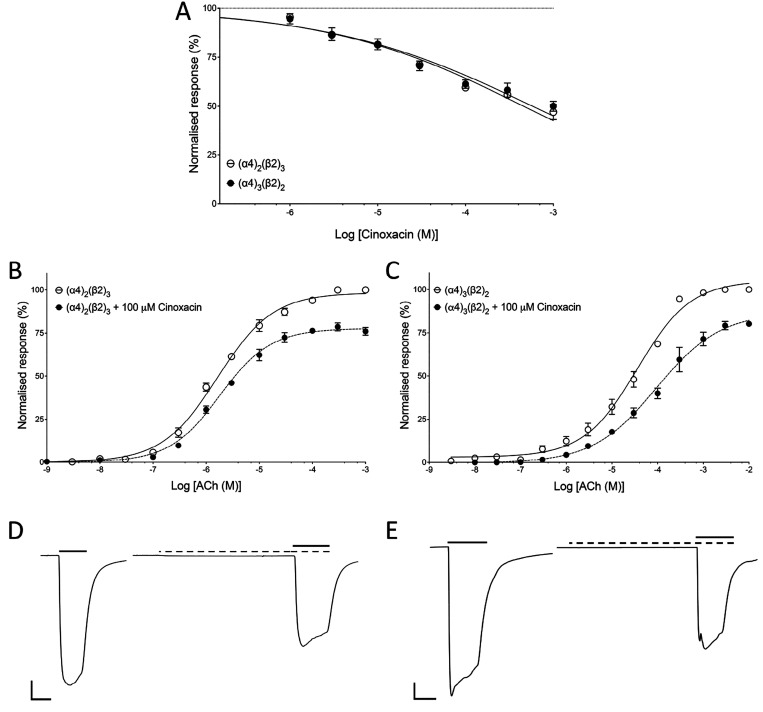
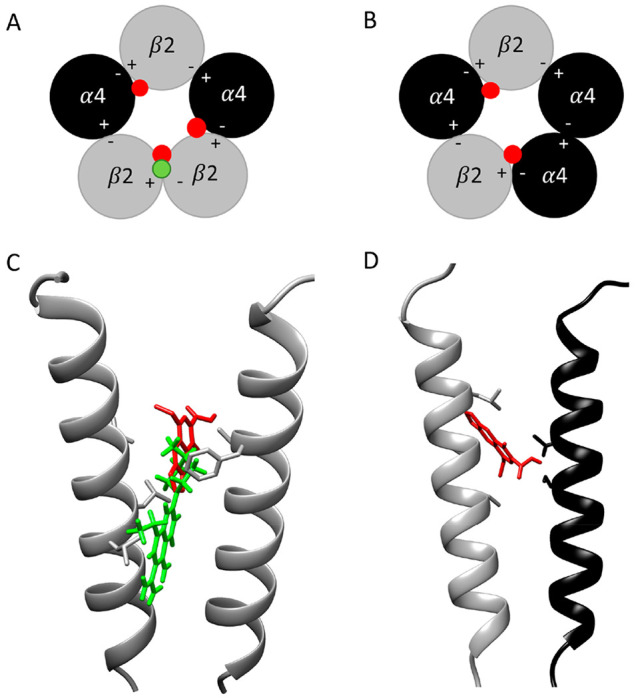
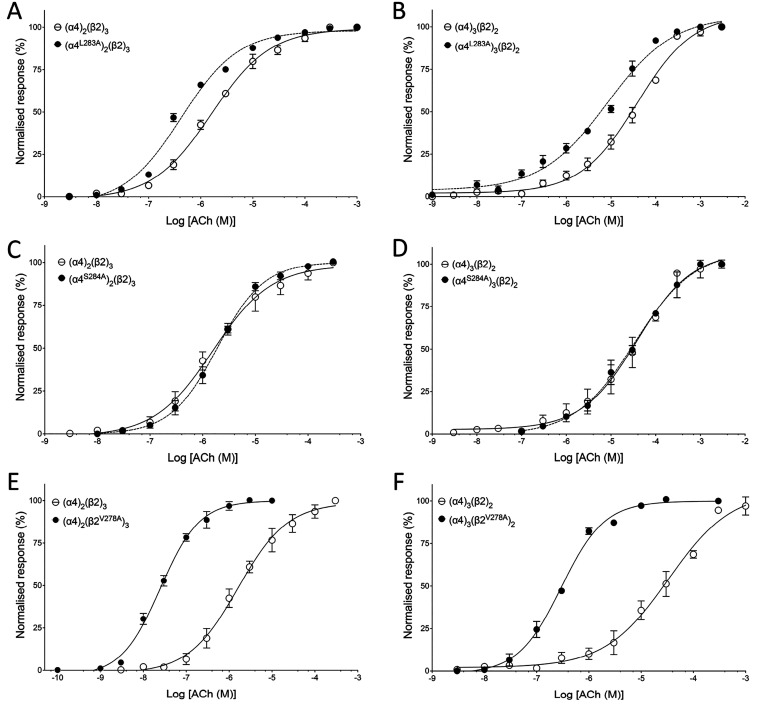
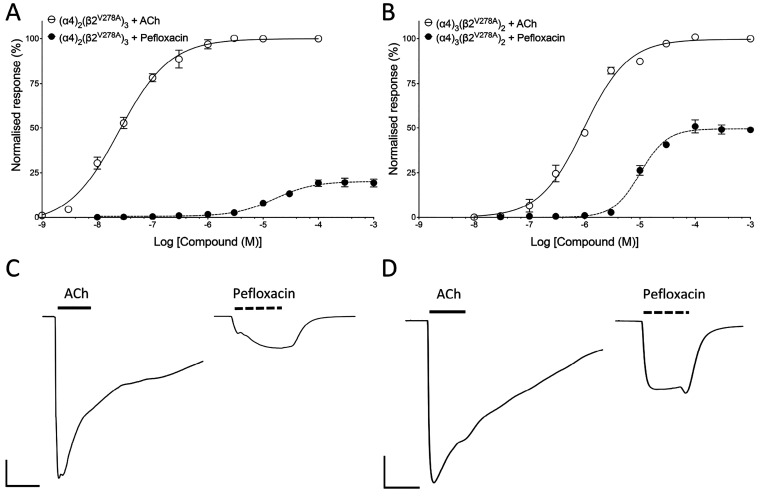
Quinolone antibiotics disrupt bacterial DNA synthesis by interacting with DNA gyrase and topoisomerase IV. However, in addition, they have been shown to act as inhibitors of pentameric ligand-gated ion channels such as GABA receptors and the α7 nicotinic acetylcholine receptor (nAChR). In the present study, we have examined the effects of quinolone antibiotics on the human α4β2 nAChR, an important subtype that is widely expressed in the central nervous system. A key feature of α4β2 nAChRs is their ability to coassemble into two distinct stoichiometries, (α4)(β2) and (α4)(β2), which results in differing affinities for acetylcholine. The effects of nine quinolone antibiotics were examined on both stoichiometries of the α4β2 receptor by two-electrode voltage-clamp recording. All compounds exhibited significant inhibition of α4β2 nAChRs. However, all of the fluoroquinolone antibiotics examined (ciprofloxacin, enoxacin, enrofloxacin, difloxacin, norfloxacin, pefloxacin, and sparfloxacin) were significantly more potent inhibitors of (α4)(β2) nAChRs than of (α4)(β2) nAChRs. This stoichiometry-selective effect was most pronounced with pefloxacin, which inhibited (α4)(β2) nAChRs with an IC of 26.4 ± 3.4 μM but displayed no significant inhibition of (α4)(β2) nAChRs. In contrast, two nonfluorinated quinolone antibiotics (cinoxacin and oxolinic acid) exhibited no selectivity in their inhibition of the two stoichiometries of α4β2. Computational docking studies suggest that pefloxacin interacts selectively with an allosteric transmembrane site at the β2(+)/β2(-) subunit interface, which is consistent with its selective inhibition of (α4)(β2). These findings concerning the antagonist effects of fluoroquinolones provide further evidence that differences in the subunit stoichiometry of heteromeric nAChRs can result in substantial differences in pharmacological properties.
喹诺酮类抗生素通过与 DNA 回旋酶和拓扑异构酶 IV 相互作用来破坏细菌 DNA 的合成。然而,除此之外,它们还被证明是五聚体配体门控离子通道(如 GABA 受体和α7 烟碱型乙酰胆碱受体(nAChR))的抑制剂。在本研究中,我们检查了喹诺酮类抗生素对人α4β2 nAChR 的影响,α4β2 nAChR 是一种广泛表达于中枢神经系统的重要亚型。α4β2 nAChR 的一个关键特征是它们能够组装成两种不同的化学计量比,(α4)(β2)和(α4)(β2),这导致对乙酰胆碱的亲和力不同。通过双电极电压钳记录,研究了 9 种喹诺酮类抗生素对α4β2 受体两种化学计量比的影响。所有化合物均显著抑制α4β2 nAChR。然而,所有测试的氟喹诺酮类抗生素(环丙沙星、恩诺沙星、恩氟沙星、二氟沙星、诺氟沙星、培氟沙星和司帕沙星)对(α4)(β2)nAChR 的抑制作用均明显强于(α4)(β2)nAChR。这种化学计量比选择性效应在培氟沙星中最为明显,其抑制(α4)(β2)nAChR 的 IC 为 26.4±3.4μM,但对(α4)(β2)nAChR 没有明显的抑制作用。相比之下,两种非氟喹诺酮类抗生素(西诺沙星和奥洛林酸)在抑制α4β2 的两种化学计量比方面没有选择性。计算对接研究表明,培氟沙星选择性地与β2(+) /β2(-)亚基界面的变构跨膜位点相互作用,这与其对(α4)(β2)的选择性抑制一致。这些关于氟喹诺酮类拮抗剂作用的发现进一步证明,异源 nAChR 的亚基化学计量比的差异可导致药理学特性的显著差异。