MRC Human Genetics Unit, Western General Hospital, Institute of Genetics and Cancer, The University of Edinburgh, Crewe Road South, Edinburgh EH4 2XU, UK.
Transforming Genetic Medicine Initiative, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, UK.
Database (Oxford). 2022 Jun 7;2022. doi: 10.1093/database/baac038.
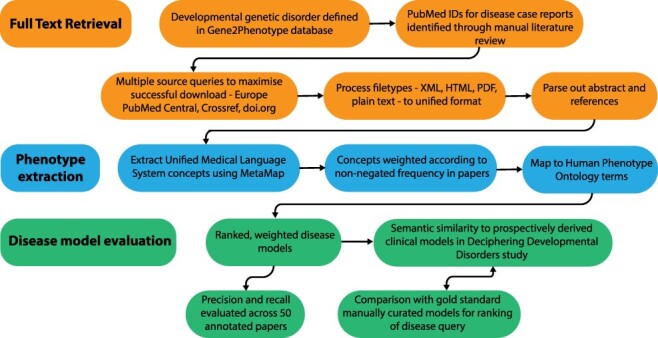
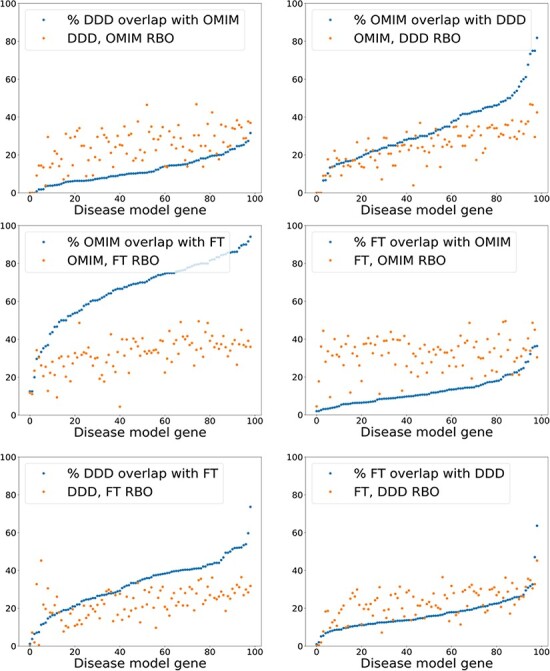
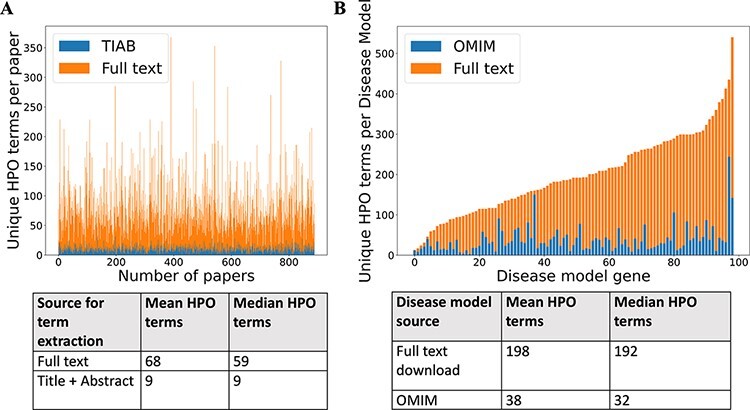
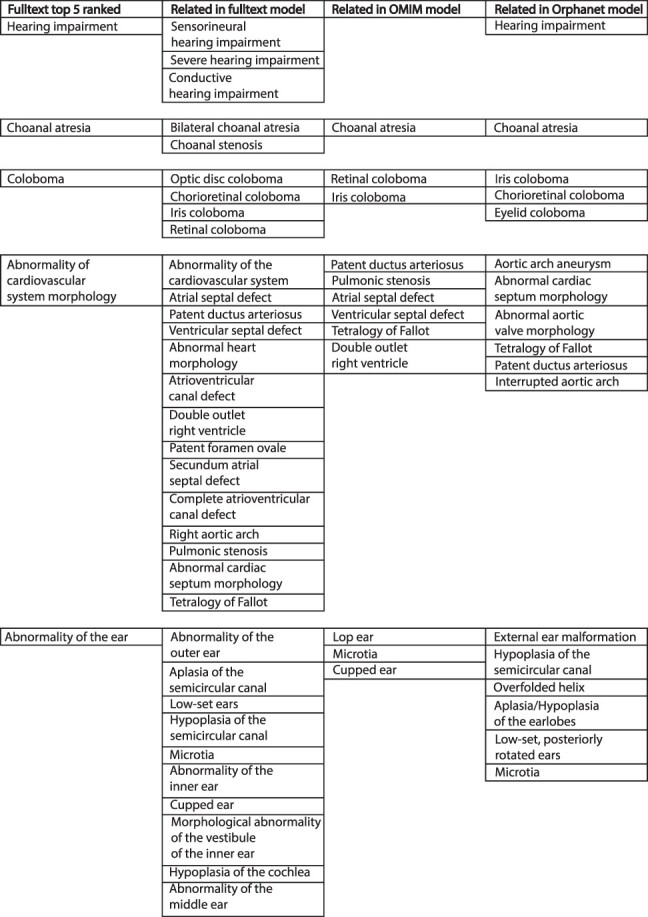
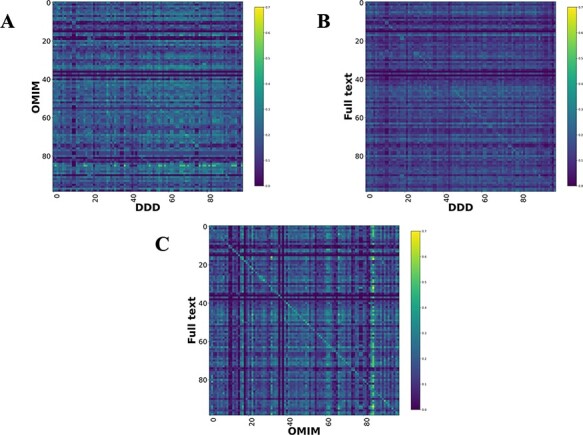
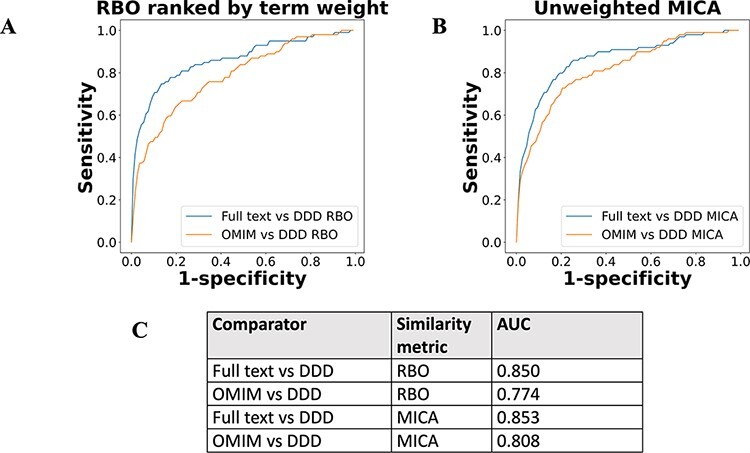
There are >2500 different genetically determined developmental disorders (DD), which, as a group, show very high levels of both locus and allelic heterogeneity. This has led to the wide-spread use of evidence-based filtering of genome-wide sequence data as a diagnostic tool in DD. Determining whether the association of a filtered variant at a specific locus is a plausible explanation of the phenotype in the proband is crucial and commonly requires extensive manual literature review by both clinical scientists and clinicians. Access to a database of weighted clinical features extracted from rigorously curated literature would increase the efficiency of this process and facilitate the development of robust phenotypic similarity metrics. However, given the large and rapidly increasing volume of published information, conventional biocuration approaches are becoming impractical. Here, we present a scalable, automated method for the extraction of categorical phenotypic descriptors from the full-text literature. Papers identified through literature review were downloaded and parsed using the Cadmus custom retrieval package. Human Phenotype Ontology terms were extracted using MetaMap, with 76-84% precision and 65-73% recall. Mean terms per paper increased from 9 in title + abstract, to 68 using full text. We demonstrate that these literature-derived disease models plausibly reflect true disease expressivity more accurately than widely used manually curated models, through comparison with prospectively gathered data from the Deciphering Developmental Disorders study. The area under the curve for receiver operating characteristic (ROC) curves increased by 5-10% through the use of literature-derived models. This work shows that scalable automated literature curation increases performance and adds weight to the need for this strategy to be integrated into informatic variant analysis pipelines. Database URL: https://doi.org/10.1093/database/baac038.
有超过 2500 种不同的遗传性发育障碍(DD),这些障碍在基因座和等位基因异质性方面都表现出非常高的水平。这导致了广泛使用基于证据的全基因组序列数据过滤作为 DD 的诊断工具。确定在特定基因座过滤的变异与表型之间的关联是否是先证者表型的合理解释至关重要,通常需要临床科学家和临床医生进行广泛的手动文献综述。访问从严格编辑的文献中提取的加权临床特征数据库将提高该过程的效率,并有助于开发稳健的表型相似性度量标准。然而,鉴于已发表信息的数量庞大且增长迅速,传统的生物注释方法变得不切实际。在这里,我们提出了一种可扩展的、自动化的从全文文献中提取分类表型描述符的方法。通过文献综述确定的论文通过 Cadmus 自定义检索包下载并解析。使用 MetaMap 提取人类表型本体论术语,具有 76-84%的精度和 65-73%的召回率。每篇论文的平均术语数从标题+摘要中的 9 个增加到使用全文时的 68 个。我们通过与 Deciphering Developmental Disorders 研究中前瞻性收集的数据进行比较,证明这些从文献中得出的疾病模型比广泛使用的手动编辑模型更能准确地反映真实的疾病表现。通过使用文献衍生模型,接收者操作特征(ROC)曲线下的面积增加了 5-10%。这项工作表明,可扩展的自动化文献注释提高了性能,并增加了将这种策略集成到信息变体分析管道中的必要性。数据库 URL:https://doi.org/10.1093/database/baac038。