Canada's Michael Smith Genome Sciences Centre, BC Cancer, Vancouver, BC, V5Z 4S6, Canada.
Department of Medical Genetics, University of British Columbia, Vancouver, BC, V5Z 4H4, Canada.
Sci Rep. 2022 Jun 7;12(1):9352. doi: 10.1038/s41598-022-13024-4.
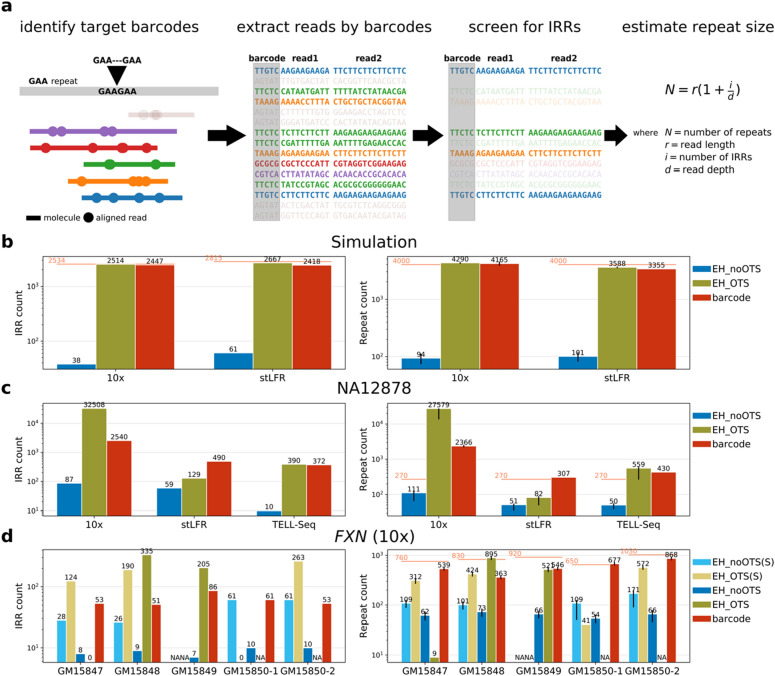
Detection of short tandem repeat (STR) expansions with standard short-read sequencing is challenging due to the difficulty in mapping multicopy repeat sequences. In this study, we explored how the long-range sequence information of barcode linked-read sequencing (BLRS) can be leveraged to improve repeat-read detection. We also devised a novel algorithm using BLRS barcodes for distance estimation and evaluated its application for STR genotyping. Both approaches were designed for genotyping large expansions (> 1 kb) that cannot be sized accurately by existing methods. Using simulated and experimental data of genomes with STR expansions from multiple BLRS platforms, we validated the utility of barcode and phasing information in attaining better STR genotypes compared to standard short-read sequencing. Although the coverage bias of extremely GC-rich STRs is an important limitation of BLRS, BLRS is an effective strategy for genotyping many other STR loci.
由于多拷贝重复序列的映射困难,使用标准短读测序检测短串联重复(STR)扩展具有挑战性。在这项研究中,我们探讨了如何利用条码连接读取测序(BLRS)的长程序列信息来提高重复读取检测。我们还设计了一种使用 BLRS 条码进行距离估计的新算法,并评估了其在 STR 基因分型中的应用。这两种方法都是为了对无法通过现有方法准确测量大小的大扩展(>1kb)进行基因分型而设计的。使用来自多个 BLRS 平台的具有 STR 扩展的基因组的模拟和实验数据,我们验证了与标准短读测序相比,条码和相位信息在获得更好的 STR 基因型方面的效用。尽管 BLRS 中极度富含 GC 的 STR 的覆盖偏差是一个重要的限制,但 BLRS 是对许多其他 STR 基因座进行基因分型的有效策略。