Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China.
Umeå Plant Science Center, Department of Forestry Genetics and Plant Physiology, Swedish University of Agricultural Sciences, Umeå, Sweden.
Nature. 2022 Jun;606(7914):527-534. doi: 10.1038/s41586-022-04808-9. Epub 2022 Jun 8.
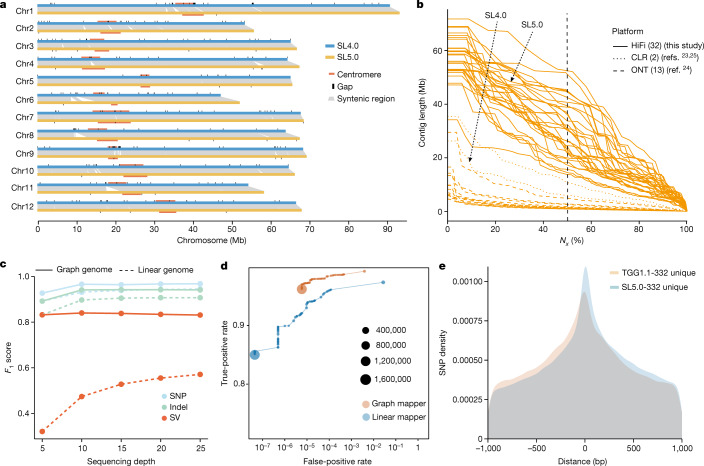
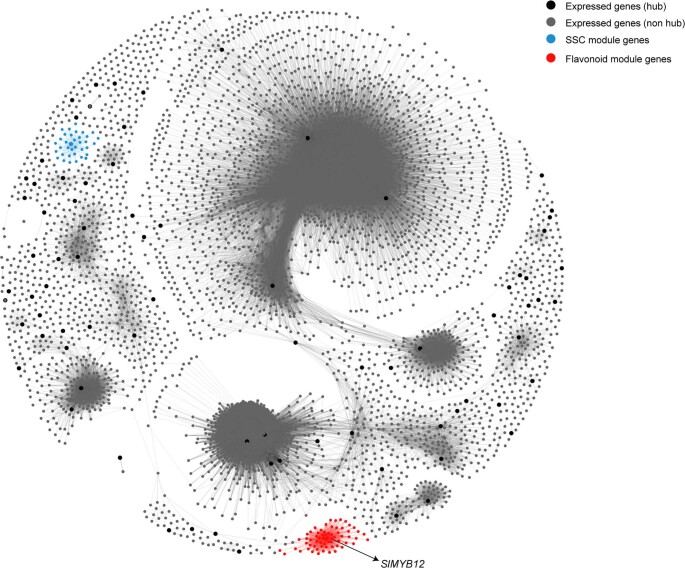
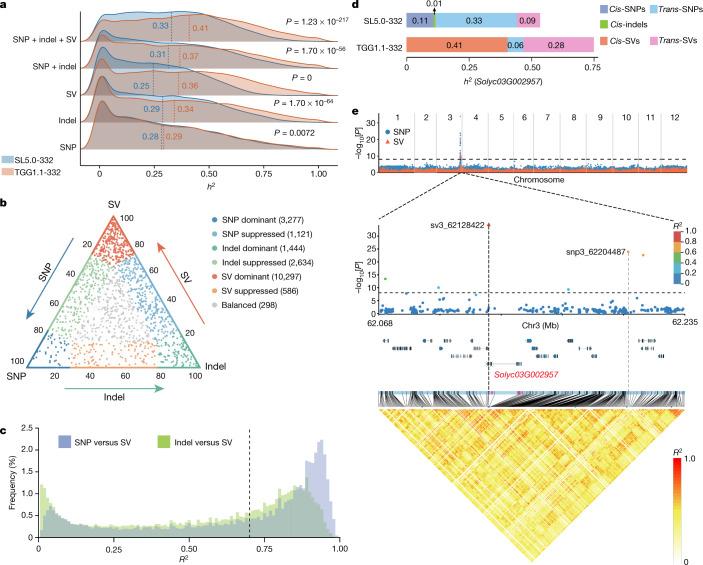
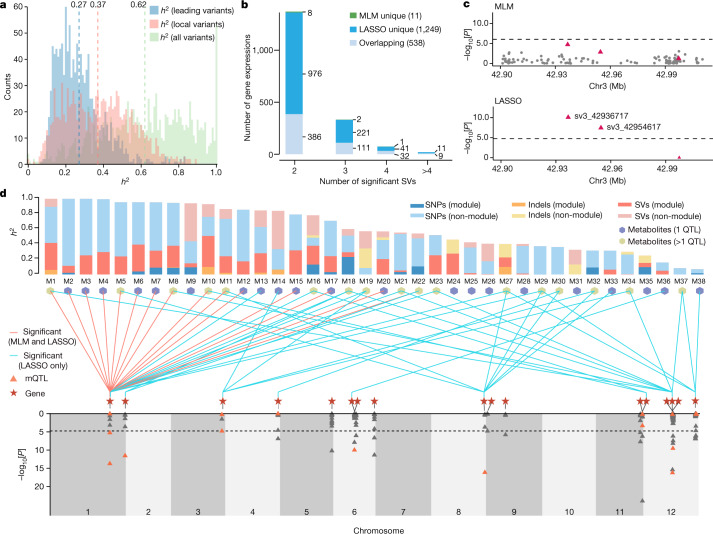
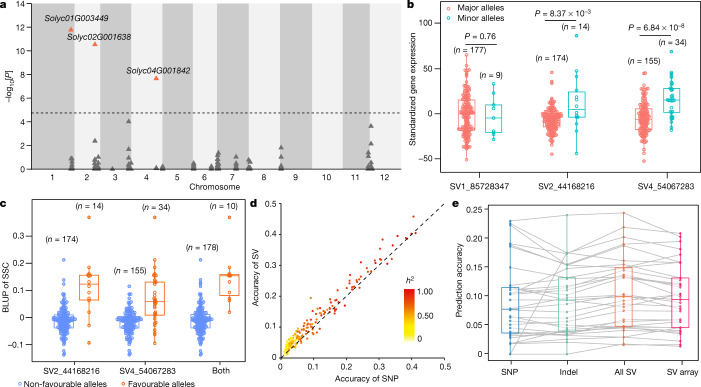
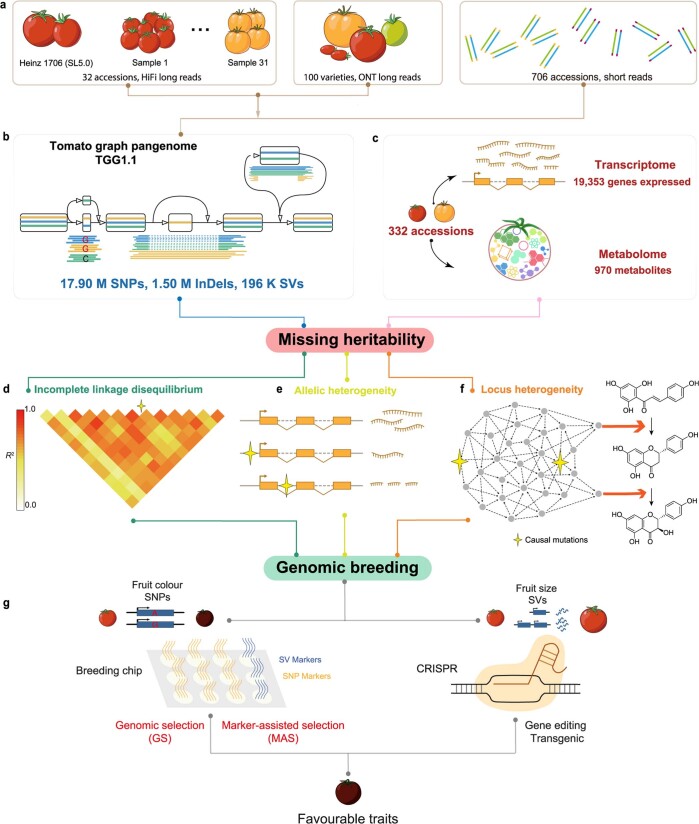
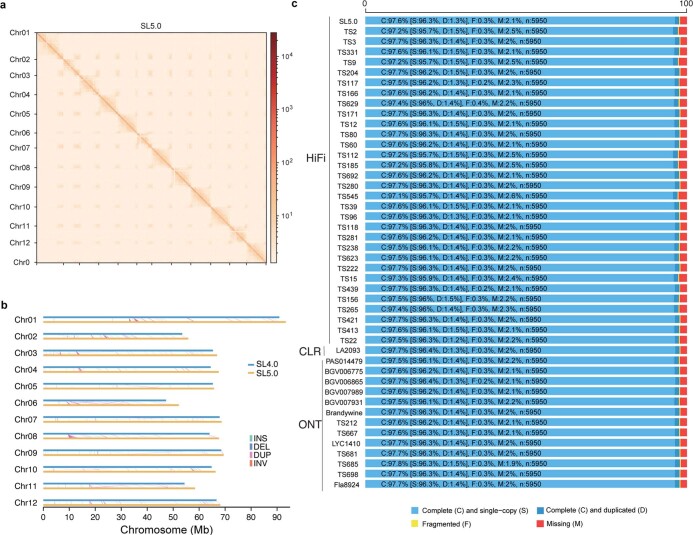
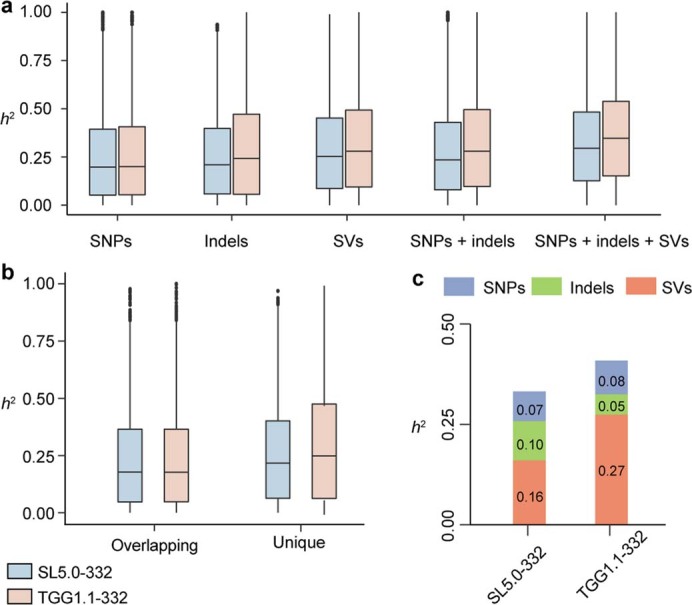
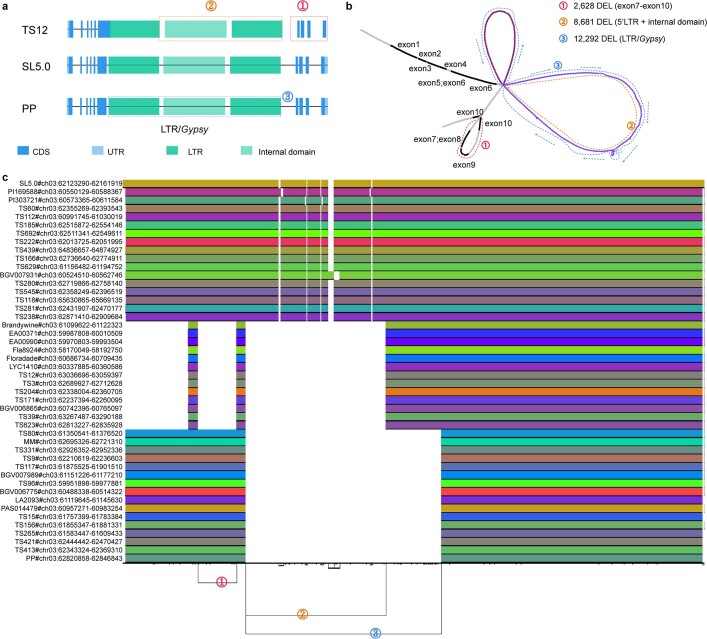
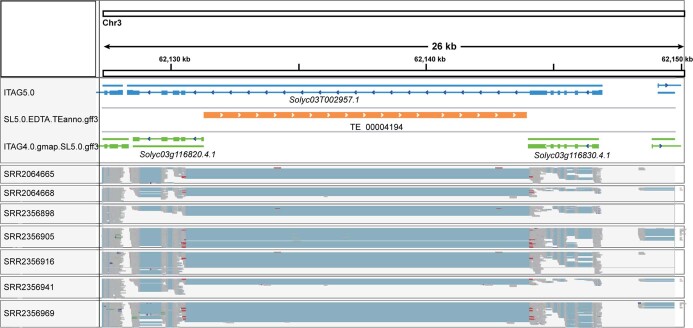
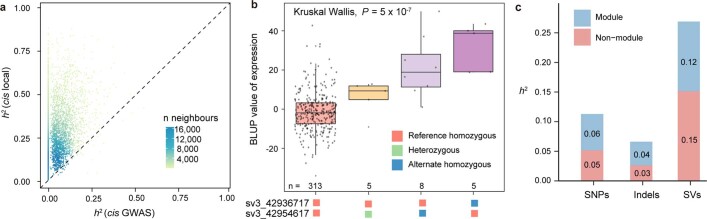
Missing heritability in genome-wide association studies defines a major problem in genetic analyses of complex biological traits. The solution to this problem is to identify all causal genetic variants and to measure their individual contributions. Here we report a graph pangenome of tomato constructed by precisely cataloguing more than 19 million variants from 838 genomes, including 32 new reference-level genome assemblies. This graph pangenome was used for genome-wide association study analyses and heritability estimation of 20,323 gene-expression and metabolite traits. The average estimated trait heritability is 0.41 compared with 0.33 when using the single linear reference genome. This 24% increase in estimated heritability is largely due to resolving incomplete linkage disequilibrium through the inclusion of additional causal structural variants identified using the graph pangenome. Moreover, by resolving allelic and locus heterogeneity, structural variants improve the power to identify genetic factors underlying agronomically important traits leading to, for example, the identification of two new genes potentially contributing to soluble solid content. The newly identified structural variants will facilitate genetic improvement of tomato through both marker-assisted selection and genomic selection. Our study advances the understanding of the heritability of complex traits and demonstrates the power of the graph pangenome in crop breeding.
全基因组关联研究中缺失的遗传力定义了遗传分析复杂生物性状的主要问题。解决这个问题的方法是识别所有的因果遗传变异,并测量它们的个体贡献。在这里,我们报告了一个番茄的图谱泛基因组,该图谱通过精确地编目了来自 838 个基因组的超过 1900 万个变异,包括 32 个新的参考水平基因组组装。该图谱泛基因组被用于 20323 个基因表达和代谢性状的全基因组关联研究分析和遗传力估计。与使用单一线性参考基因组相比,平均估计性状遗传力为 0.41。这种 24%的遗传力估计增加主要是由于通过包含使用图谱泛基因组识别的额外因果结构变异来解决不完全连锁不平衡。此外,通过解决等位基因和基因座异质性,结构变异提高了识别对农业重要性状有潜在遗传因素的能力,例如,确定了两个可能有助于可溶性固形物含量的新基因。新鉴定的结构变异将通过标记辅助选择和基因组选择促进番茄的遗传改良。我们的研究推进了对复杂性状遗传力的理解,并展示了图谱泛基因组在作物育种中的强大功能。