Quan Cheng, Lu Hao, Lu Yiming, Zhou Gangqiao
Department of Genetics & Integrative Omics, State Key Laboratory of Proteomics, National Center for Protein Sciences, Beijing Institute of Radiation Medicine, Beijing 100850, PR China.
Hebei University, Baoding, Hebei Province 071002, PR China.
Comput Struct Biotechnol J. 2022 May 27;20:2639-2647. doi: 10.1016/j.csbj.2022.05.047. eCollection 2022.
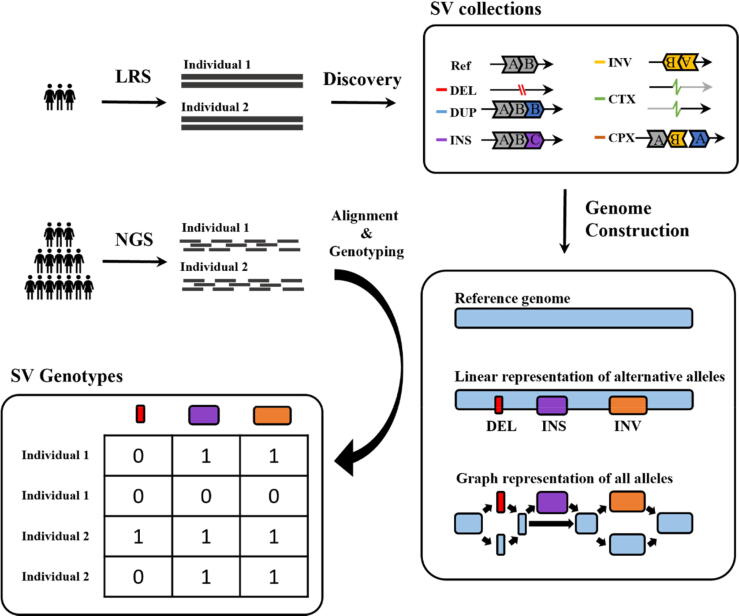
Population-scale studies of structural variation (SV) are growing rapidly worldwide with the development of long-read sequencing technology, yielding a considerable number of novel SVs and complete gap-closed genome assemblies. Herein, we highlight recent studies using a hybrid sequencing strategy and present the challenges toward large-scale genotyping for SVs due to the reference bias. Genotyping SVs at a population scale remains challenging, which severely impacts genotype-based population genetic studies or genome-wide association studies of complex diseases. We summarize academic efforts to improve genotype quality through linear or graph representations of reference and alternative alleles. Graph-based genotypers capable of integrating diverse genetic information are effectively applied to large and diverse cohorts, contributing to unbiased downstream analysis. Meanwhile, there is still an urgent need in this field for efficient tools to construct complex graphs and perform sequence-to-graph alignments.
随着长读长测序技术的发展,全球范围内针对结构变异(SV)的群体规模研究正在迅速增加,产生了大量新的SV以及完整的缺口封闭基因组组装。在此,我们重点介绍了近期使用混合测序策略的研究,并指出了由于参考偏差导致的大规模SV基因分型所面临的挑战。在群体规模上对SV进行基因分型仍然具有挑战性,这严重影响了基于基因型的群体遗传学研究或复杂疾病的全基因组关联研究。我们总结了通过参考等位基因和替代等位基因的线性或图形表示来提高基因型质量的学术努力。能够整合多种遗传信息的基于图形的基因分型器被有效地应用于大型多样的队列研究,有助于进行无偏的下游分析。与此同时,该领域仍然迫切需要高效的工具来构建复杂图形并进行序列到图形的比对。