D. E. Shaw Research, New York, New York 10036, United States.
Department of Biochemistry and Molecular Biophysics, Columbia University, New York, New York 10032, United States.
J Phys Chem B. 2022 Jun 23;126(24):4442-4457. doi: 10.1021/acs.jpcb.1c10971. Epub 2022 Jun 12.
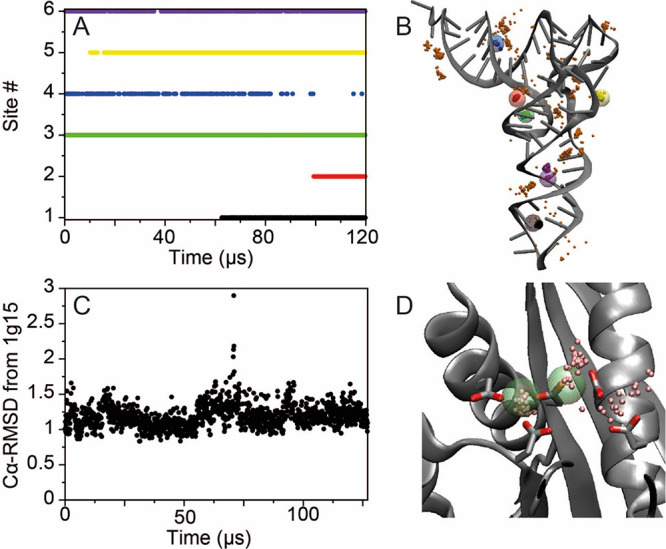
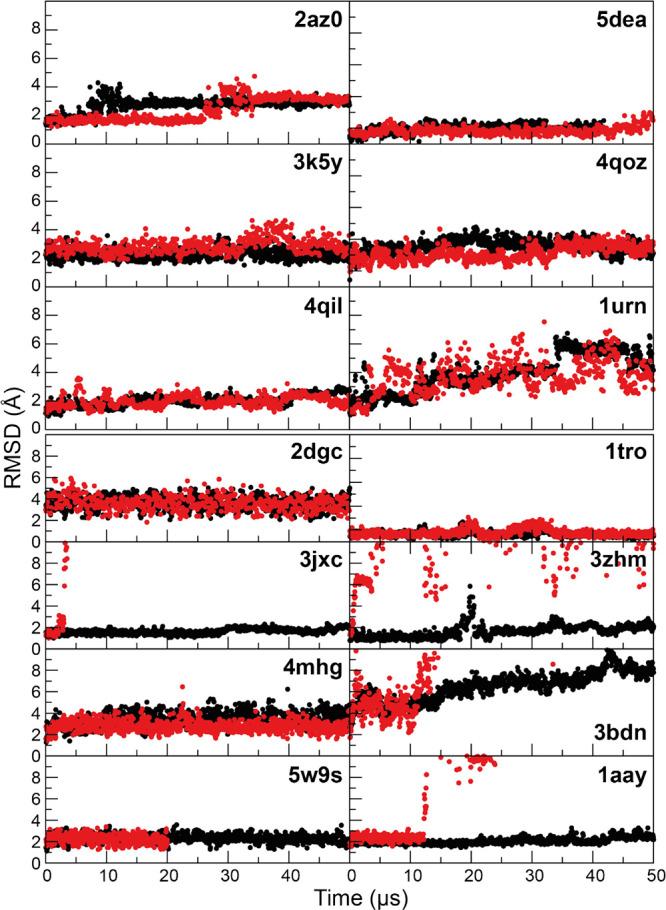
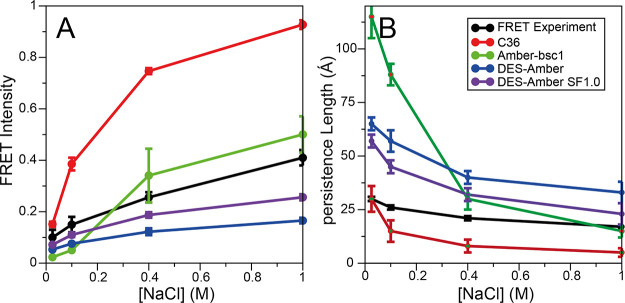
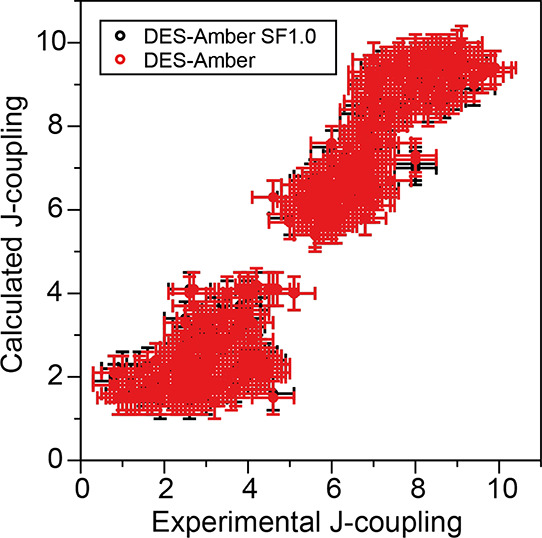
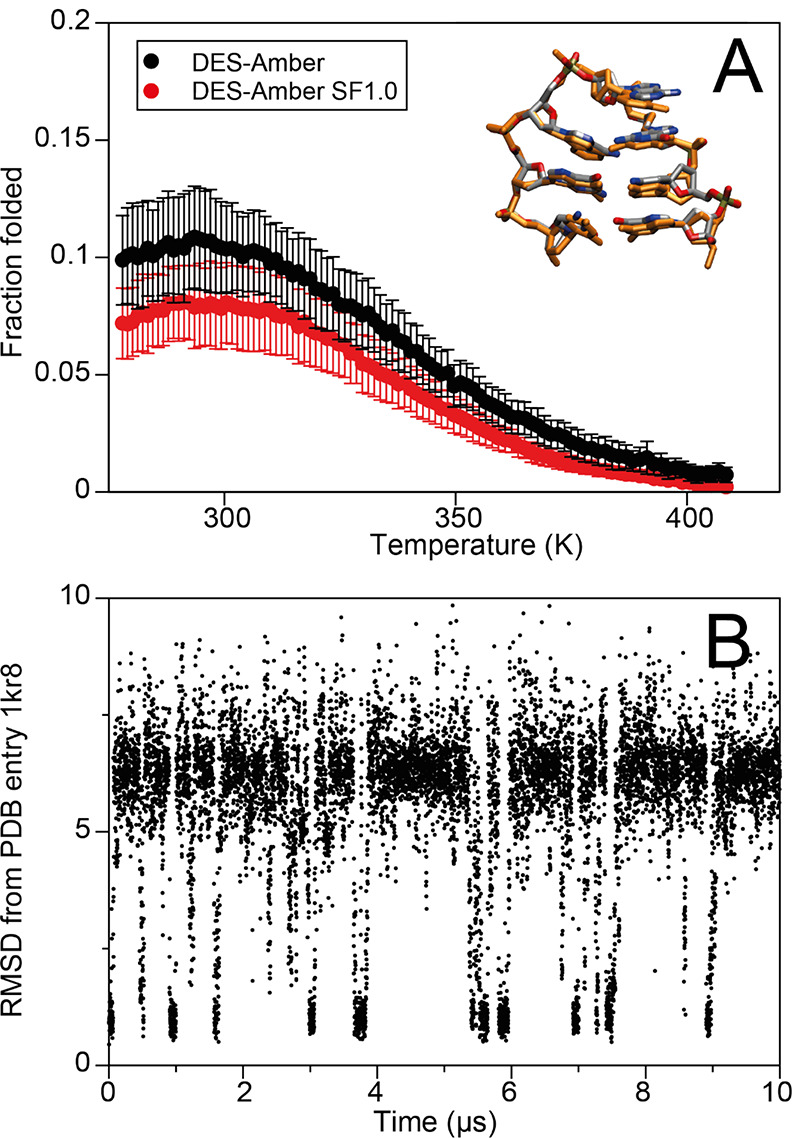
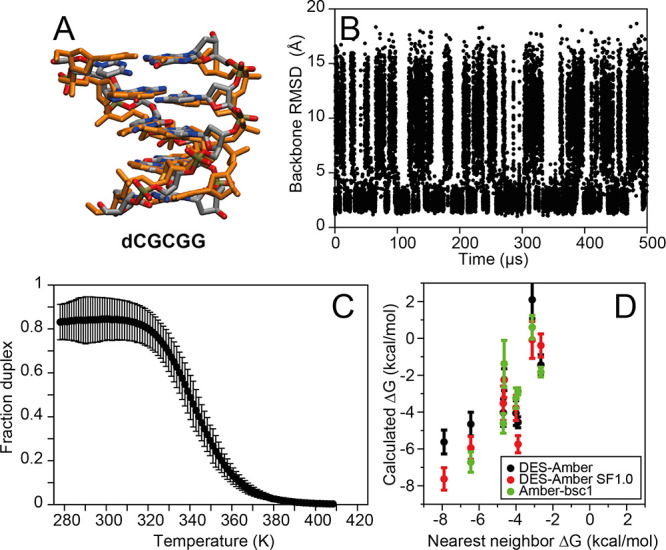
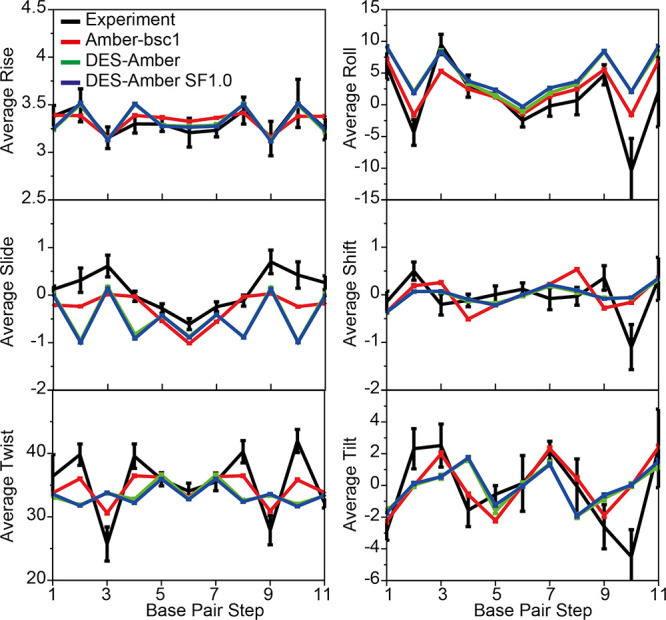
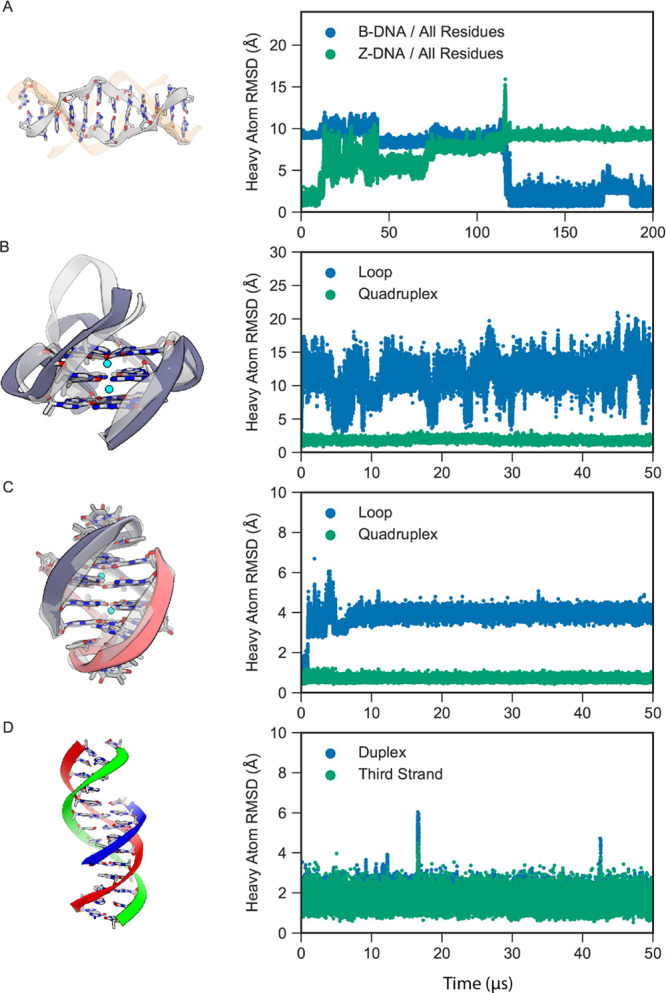
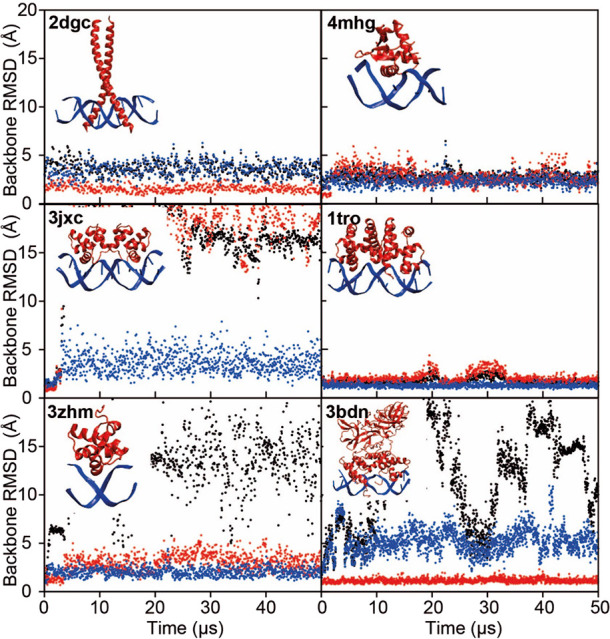
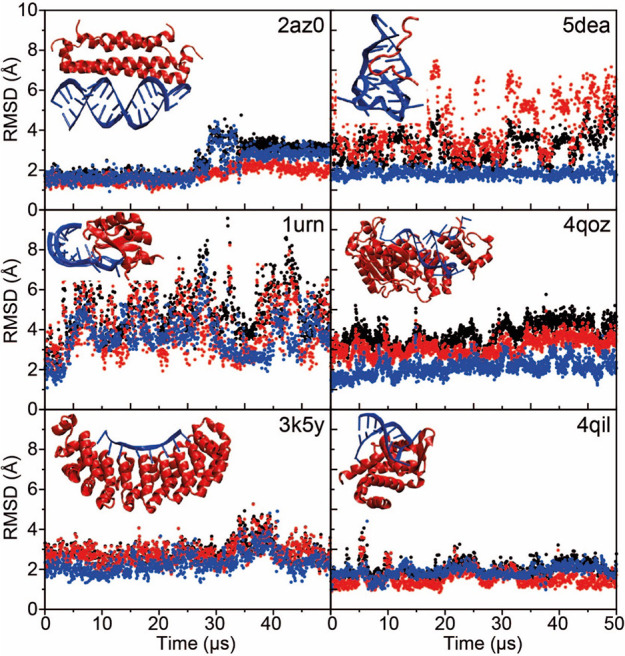
Although molecular dynamics (MD) simulations have been used extensively to study the structural dynamics of proteins, the role of MD simulation in studies of nucleic acid based systems has been more limited. One contributing factor to this disparity is the historically lower level of accuracy of the physical models used in such simulations to describe interactions involving nucleic acids. By modifying nonbonded and torsion parameters of a force field from the Amber family of models, we recently developed force field parameters for RNA that achieve a level of accuracy comparable to that of state-of-the-art protein force fields. Here we report force field parameters for DNA, which we developed by transferring nonbonded parameters from our recently reported RNA force field and making subsequent adjustments to torsion parameters. We have also modified the backbone charges in both the RNA and DNA parameter sets to make the treatment of electrostatics compatible with our recently developed variant of the Amber protein and ion force field. We name the force field resulting from the union of these three parameter sets (the new DNA parameters, the revised RNA parameters, and the existing protein and ion parameters) . Extensive testing of DES-Amber indicates that it can describe the thermal stability and conformational flexibility of single- and double-stranded DNA systems with a level of accuracy comparable to or, especially for disordered systems, exceeding that of state-of-the-art nucleic acid force fields. Finally, we show that, in certain favorable cases, DES-Amber can be used for long-timescale simulations of protein-nucleic acid complexes.
尽管分子动力学(MD)模拟已被广泛用于研究蛋白质的结构动力学,但 MD 模拟在基于核酸的系统研究中的应用更为有限。造成这种差异的一个因素是,在这些模拟中,用于描述涉及核酸的相互作用的物理模型的历史准确性较低。通过修改 Amber 模型系列中力场的非键和扭转参数,我们最近开发了 RNA 的力场参数,其准确性可与最先进的蛋白质力场相媲美。在这里,我们报告了 DNA 的力场参数,我们通过从我们最近报道的 RNA 力场中转移非键参数并对扭转参数进行后续调整来开发这些参数。我们还修改了 RNA 和 DNA 参数集中的骨架电荷,以使静电处理与我们最近开发的 Amber 蛋白质和离子力场的变体兼容。我们将这三个参数集(新的 DNA 参数、修订的 RNA 参数以及现有的蛋白质和离子参数)组合在一起的力场命名为. 通过对 DES-Amber 的广泛测试表明,它可以描述单链和双链 DNA 系统的热稳定性和构象灵活性,其准确性可与最先进的核酸力场相媲美,尤其是对于无序系统,甚至超过了最先进的核酸力场。最后,我们表明,在某些有利情况下,DES-Amber 可用于蛋白质-核酸复合物的长时间尺度模拟。