Department of Biostatistics, Yale School of Public Health, 60 College Street, New Haven, USA.
Department of Statistics, Sungkyunkwan University, 25-2, Sungkyunkwan-ro, Jongno-gu, Seoul, South Korea.
Genome Biol. 2022 Jun 15;23(1):129. doi: 10.1186/s13059-022-02688-w.
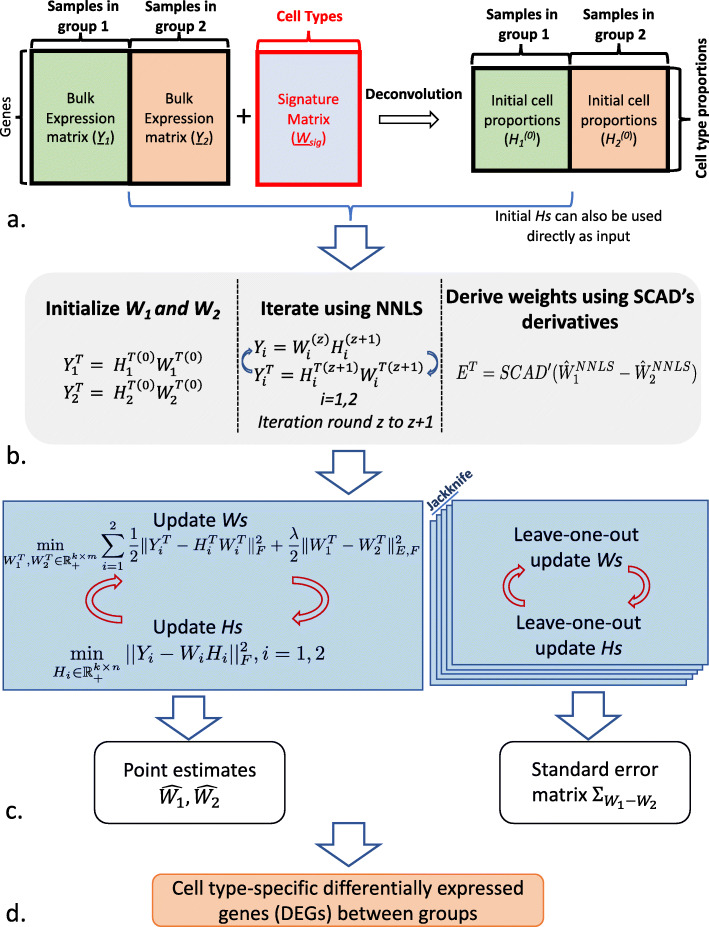
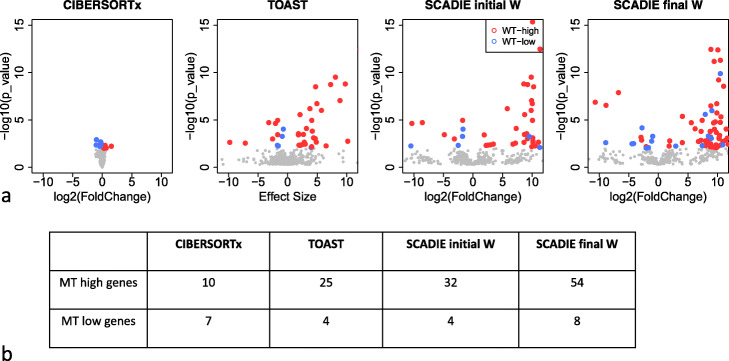
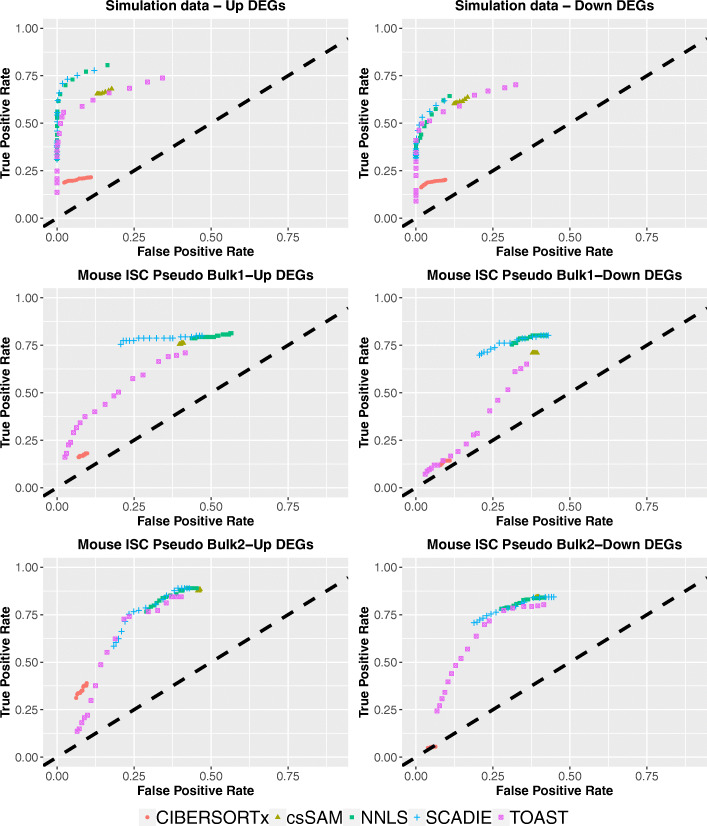
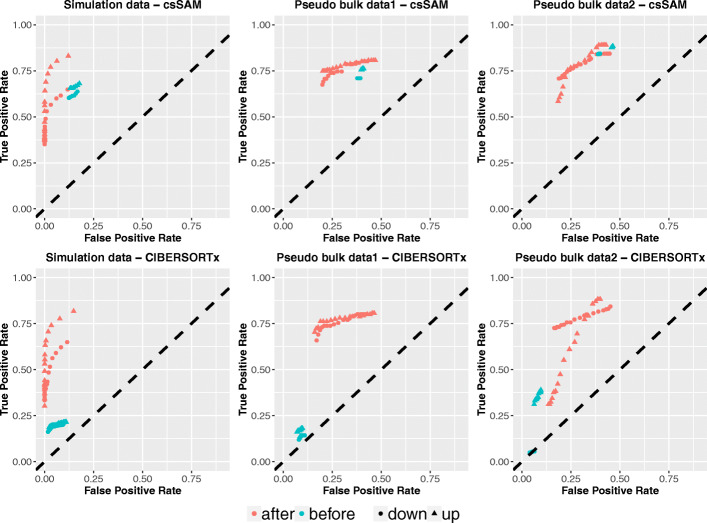
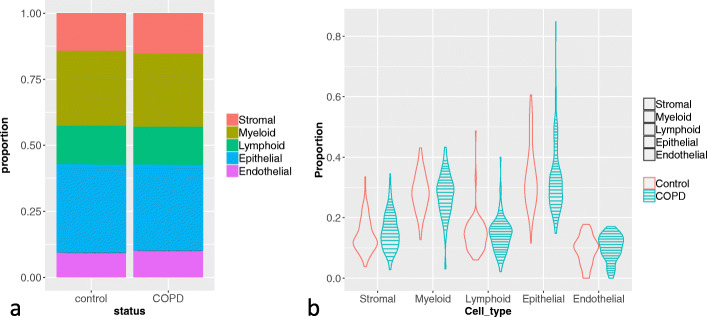
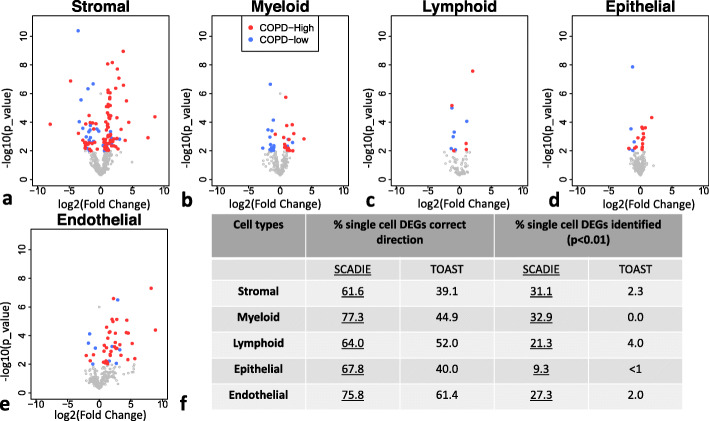
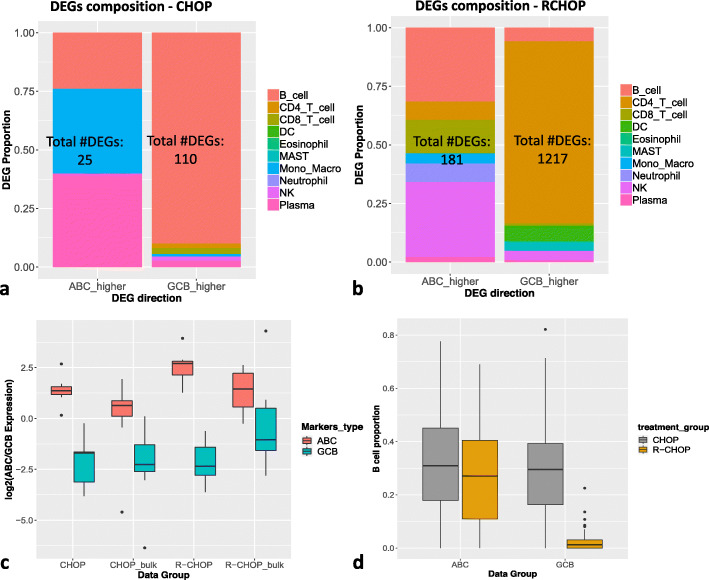
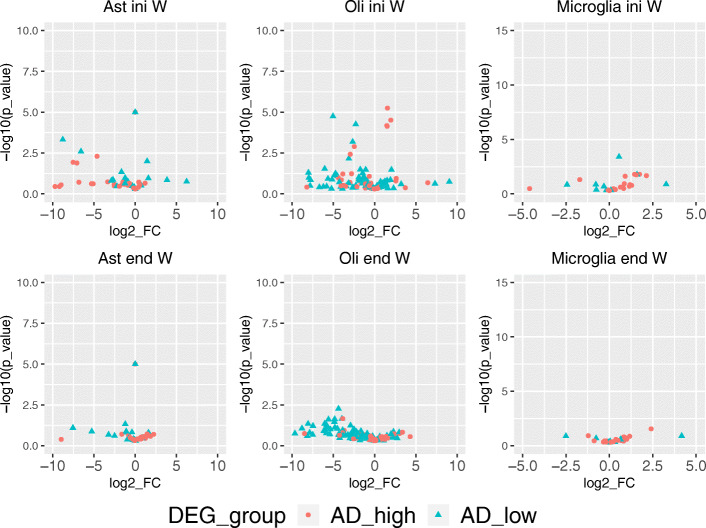
A challenge in bulk gene differential expression analysis is to differentiate changes due to cell type-specific gene expression and cell type proportions. SCADIE is an iterative algorithm that simultaneously estimates cell type-specific gene expression profiles and cell type proportions, and performs cell type-specific differential expression analysis at the group level. Through its unique penalty and objective function, SCADIE more accurately identifies cell type-specific differentially expressed genes than existing methods, including those that may be missed from single cell RNA-Seq data. SCADIE has robust performance with respect to the choice of deconvolution methods and the sources and quality of input data.
批量基因差异表达分析面临的一个挑战是区分细胞类型特异性基因表达和细胞类型比例引起的变化。SCADIE 是一种迭代算法,可同时估计细胞类型特异性基因表达谱和细胞类型比例,并在组水平上进行细胞类型特异性差异表达分析。通过其独特的惩罚和目标函数,SCADIE 比现有的方法更准确地识别细胞类型特异性差异表达基因,包括那些可能从单细胞 RNA-Seq 数据中遗漏的基因。SCADIE 对于去卷积方法的选择以及输入数据的来源和质量具有稳健的性能。