Texas A&M Drug Discovery Laboratory, Department of Chemistry, Texas A&M University, College Station, TX, 77843, USA.
Sorrento Therapeutics, Inc. San Diego, CA, 92121, USA.
Eur J Med Chem. 2022 Oct 5;240:114570. doi: 10.1016/j.ejmech.2022.114570. Epub 2022 Jun 27.
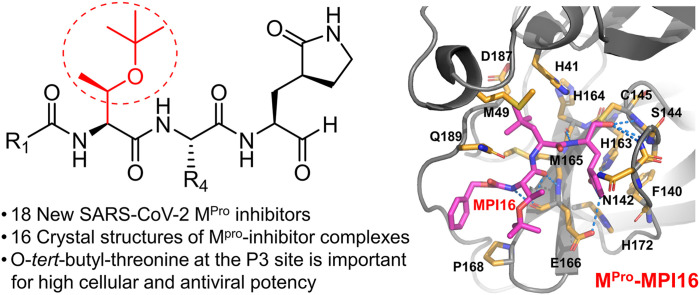
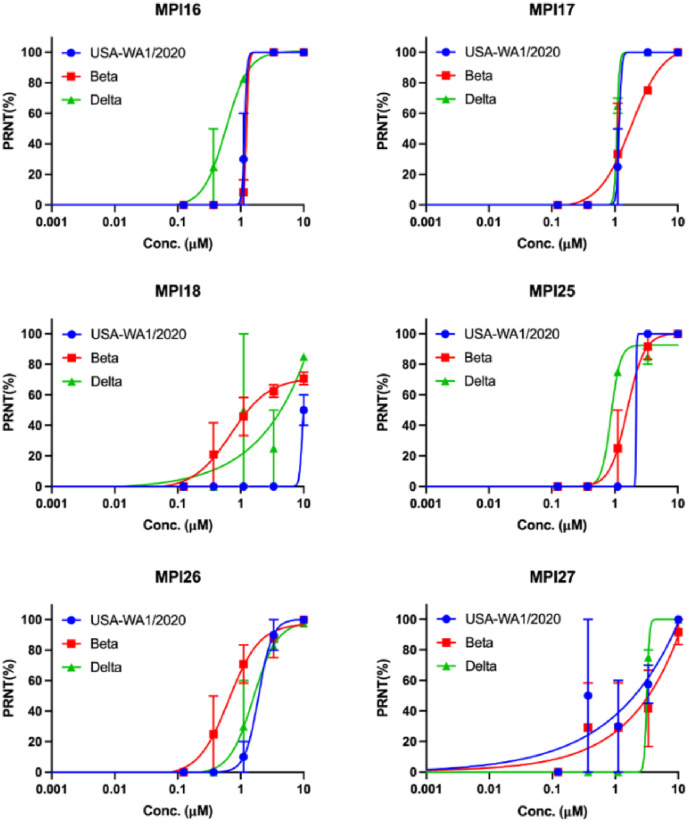
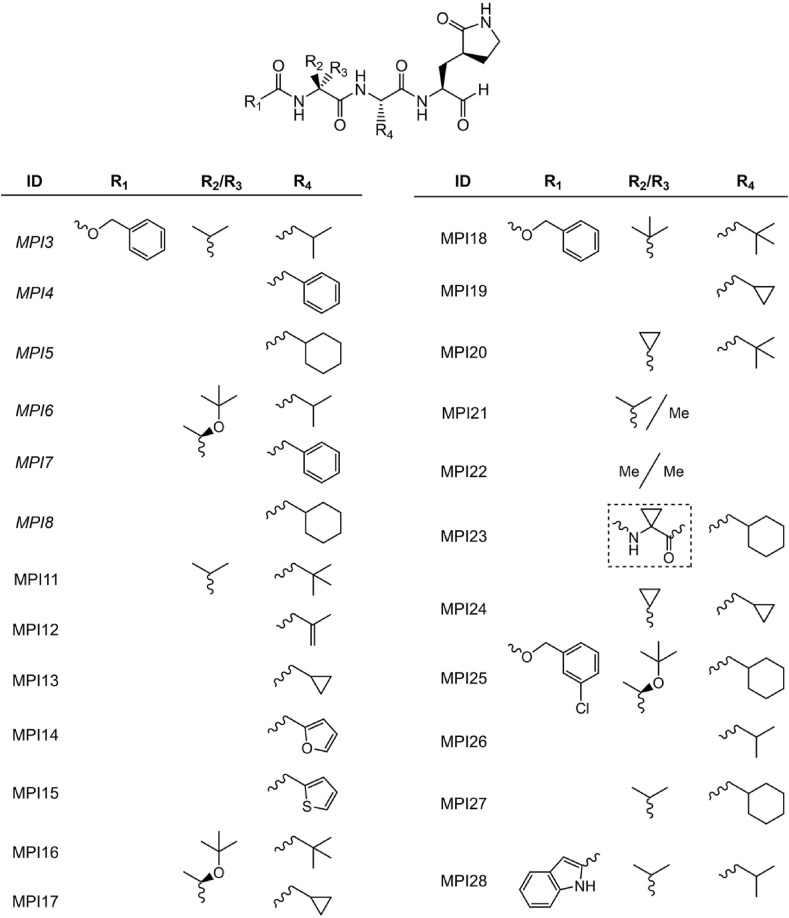
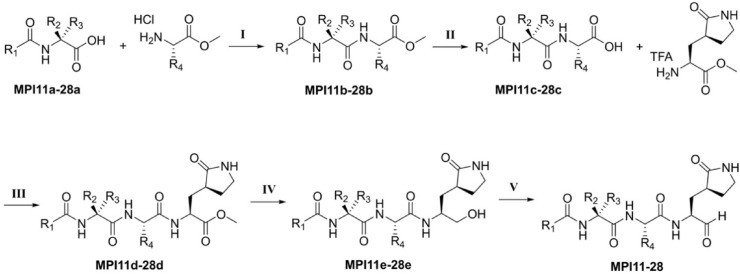
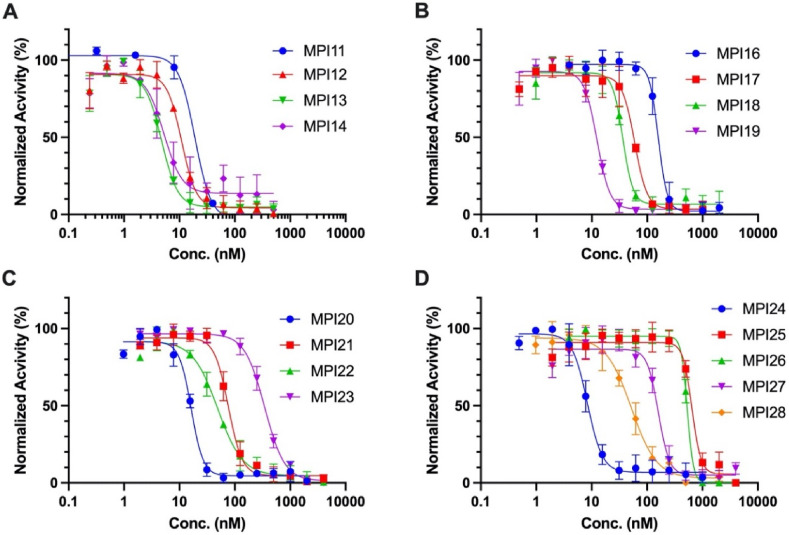
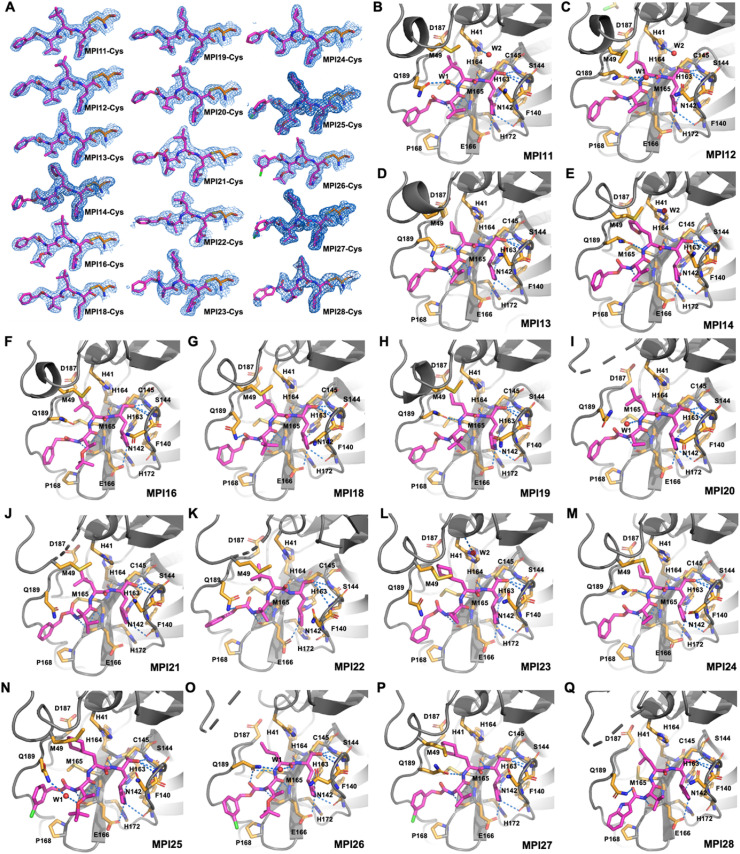
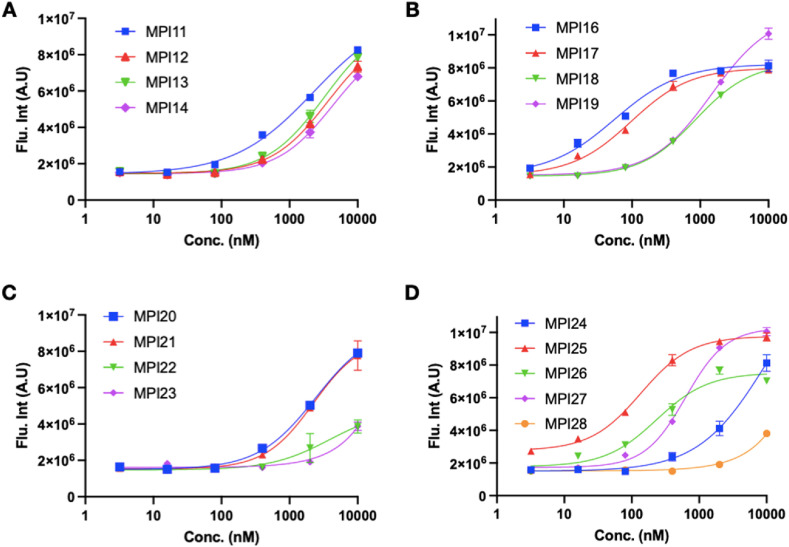
As an essential enzyme of SARS-CoV-2, the COVID-19 pathogen, main protease (M) is a viable target to develop antivirals for the treatment of COVID-19. By varying chemical compositions at both P2 and P3 positions and the N-terminal protection group, we synthesized 18 tripeptidyl M inhibitors that contained also an aldehyde warhead and β-(S-2-oxopyrrolidin-3-yl)-alaninal at the P1 position. Systematic characterizations of these inhibitors were conducted, including their in vitro enzymatic inhibition potency, X-ray crystal structures of their complexes with M, their inhibition of M transiently expressed in 293T cells, and cellular toxicity and SARS-CoV-2 antiviral potency of selected inhibitors. These inhibitors have a large variation of determined in vitro enzymatic inhibition IC values that range from 4.8 to 650 nM. The determined in vitro enzymatic inhibition IC values reveal that relatively small side chains at both P2 and P3 positions are favorable for achieving high in vitro M inhibition potency, the P3 position is tolerable toward unnatural amino acids with two alkyl substituents on the α-carbon, and the inhibition potency is sensitive toward the N-terminal protection group. X-ray crystal structures of M bound with 16 inhibitors were determined. In all structures, the M active site cysteine interacts covalently with the aldehyde warhead of the bound inhibitor to form a hemithioacetal that takes an S configuration. For all inhibitors, election density around the N-terminal protection group is weak indicating possible flexible binding of this group to M. In M, large structural variations were observed on residues N142 and Q189. Unlike their high in vitro enzymatic inhibition potency, most inhibitors showed low potency to inhibit M that was transiently expressed in 293T cells. Inhibitors that showed high potency to inhibit M transiently expressed in 293T cells all contain O-tert-butyl-threonine at the P3 position. These inhibitors also exhibited relatively low cytotoxicity and high antiviral potency. Overall, our current and previous studies indicate that O-tert-butyl-threonine at the P3 site is a key component to achieve high cellular and antiviral potency for tripeptidyl aldehyde inhibitors of M.
作为 SARS-CoV-2(引发 COVID-19 的病毒)的必需酶,新冠病毒主蛋白酶(Mpro)是开发治疗 COVID-19 的抗病毒药物的一个可行靶点。通过改变 P2 和 P3 位以及 N-端保护基团的化学组成,我们合成了 18 种包含醛基弹头和 P1 位β-(S-2-氧代吡咯烷-3-基)-丙氨酸的三肽 Mpro 抑制剂。对这些抑制剂进行了系统的表征,包括它们的体外酶抑制活性、与 Mpro 复合物的 X 射线晶体结构、它们对 293T 细胞中转瞬即逝表达的 Mpro 的抑制作用,以及选定抑制剂的细胞毒性和抗 SARS-CoV-2 活性。这些抑制剂的体外酶抑制 IC 值有很大的变化,范围从 4.8 到 650 nM。体外酶抑制 IC 值的测定表明,P2 和 P3 位的相对小侧链有利于获得高体外 Mpro 抑制活性,P3 位对α-碳上有两个烷基取代基的非天然氨基酸具有耐受性,抑制活性对 N-端保护基团敏感。测定了 16 种 Mpro 结合抑制剂的 Mpro 结合 X 射线晶体结构。在所有结构中,Mpro 活性位点半胱氨酸与结合抑制剂的醛基弹头共价相互作用,形成一个 S 构型的半硫缩醛。对于所有抑制剂,N-端保护基团周围的电子密度较弱,表明该基团与 Mpro 可能具有柔性结合。在 Mpro 中,观察到 N142 和 Q189 残基的结构变化较大。与它们的高体外酶抑制活性不同,大多数抑制剂对瞬时表达在 293T 细胞中的 Mpro 抑制活性较低。对瞬时表达在 293T 细胞中的 Mpro 具有高抑制活性的抑制剂在 P3 位都含有 O-叔丁基-苏氨酸。这些抑制剂还表现出相对较低的细胞毒性和较高的抗病毒活性。总的来说,我们目前和以前的研究表明,P3 位的 O-叔丁基-苏氨酸是实现三肽醛基 Mpro 抑制剂高细胞和抗病毒活性的关键组成部分。