Feng Cong, Wang Xingwei, Wu Shishi, Ning Weidong, Song Bo, Yan Jianbin, Cheng Shifeng
Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences (CAAS), Shenzhen, China.
State Key Laboratory of Crop Stress Adaptation and Improvement, School of Life Sciences, Henan University, Kaifeng, China.
Front Plant Sci. 2022 Jul 1;13:927407. doi: 10.3389/fpls.2022.927407. eCollection 2022.
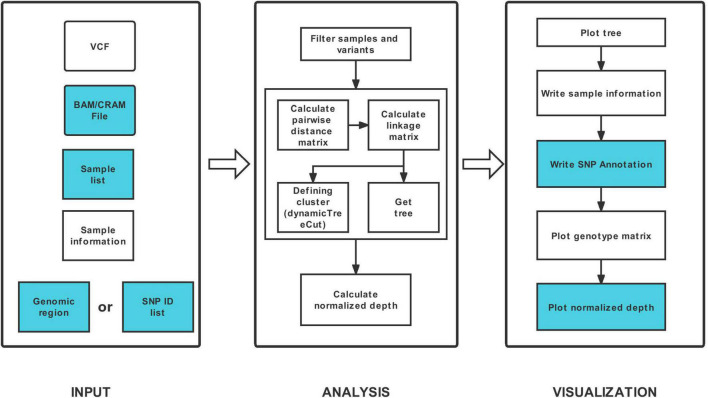
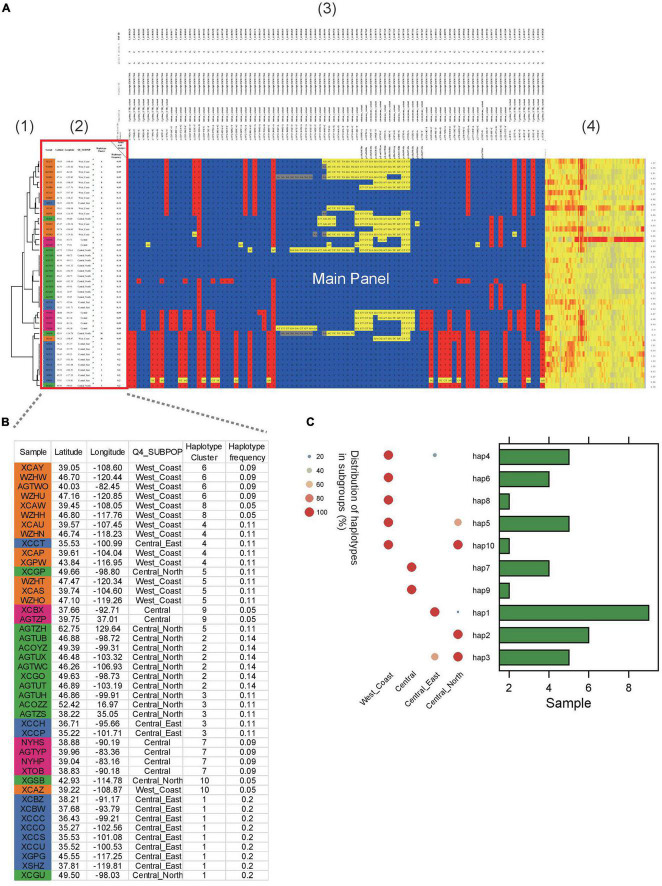
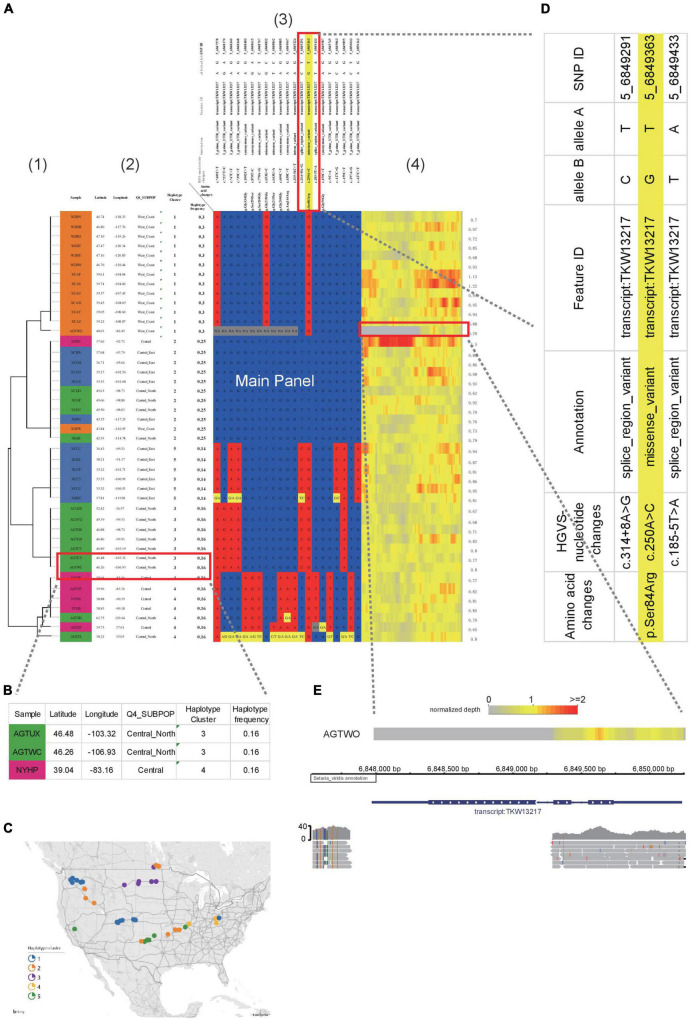
Haplotype identification, characterization and visualization are important for large-scale analysis and use in population genomics. Many tools have been developed to visualize haplotypes, but it is challenging to display both the pattern of haplotypes and the genotypes for each single SNP in the context of a large amount of genomic data. Here, we describe the tool HAPPE, which uses the agglomerative hierarchical clustering algorithm to characterize and visualize the genotypes and haplotypes in a phylogenetic context. The tool displays the plots by coloring the cells and/or their borders in Excel tables for any given gene and genomic region of interest. HAPPE facilitates informative displays wherein data in plots are easy to read and access. It allows parallel display of several lines of values, such as phylogenetic trees, values of GWAS, the entry of genes or SNPs, and the sequencing depth at each position. These features are informative for the detection of insertion/deletions or copy number variations. Overall, HAPPE provides editable plots consisting of cells in Excel tables, which are user-friendly to non-programmers. This pipeline is coded in Python and is available at https://github.com/fengcong3/HAPPE.
单倍型识别、特征描述及可视化对于群体基因组学中的大规模分析及应用而言至关重要。已经开发出许多工具用于单倍型可视化,但是在大量基因组数据背景下,既要展示单倍型模式又要展示每个单核苷酸多态性(SNP)的基因型具有挑战性。在此,我们描述了工具HAPPE,它使用凝聚层次聚类算法在系统发育背景下对基因型和单倍型进行特征描述及可视化。该工具通过为任何给定的感兴趣基因和基因组区域在Excel表格中对单元格和/或其边框进行着色来显示图表。HAPPE有助于进行信息丰富的展示,其中图表中的数据易于阅读和获取。它允许并行显示几行值,例如系统发育树、全基因组关联研究(GWAS)值、基因或SNP条目以及每个位置的测序深度。这些特征对于检测插入/缺失或拷贝数变异具有参考价值。总体而言,HAPPE提供由Excel表格中的单元格组成的可编辑图表,对非程序员来说用户友好。此流程用Python编码,可在https://github.com/fengcong3/HAPPE获取。