Department of Computer Science, Purdue University, West Lafayette, Indiana.
Department of Biological Sciences, Purdue University, West Lafayette, Indiana.
Curr Protoc. 2022 Jul;2(7):e494. doi: 10.1002/cpz1.494.
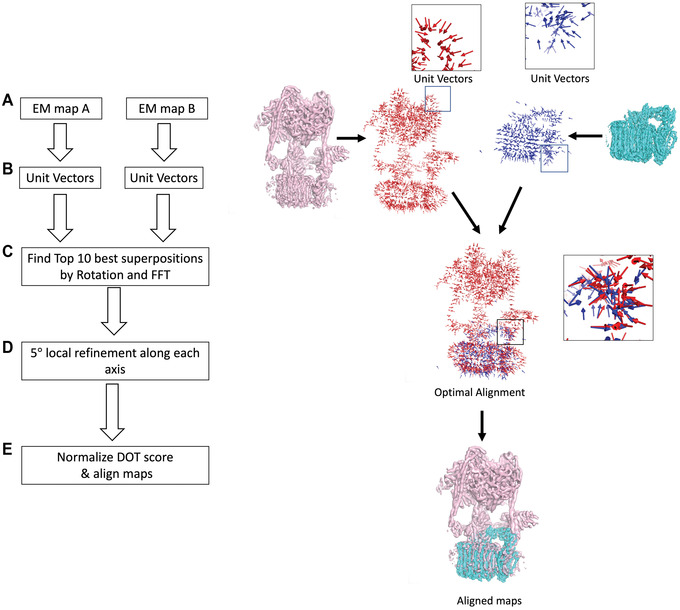
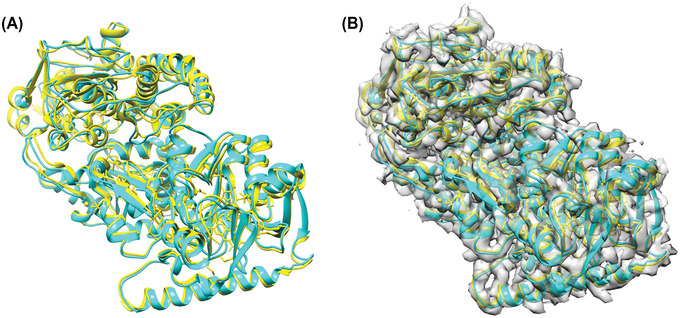
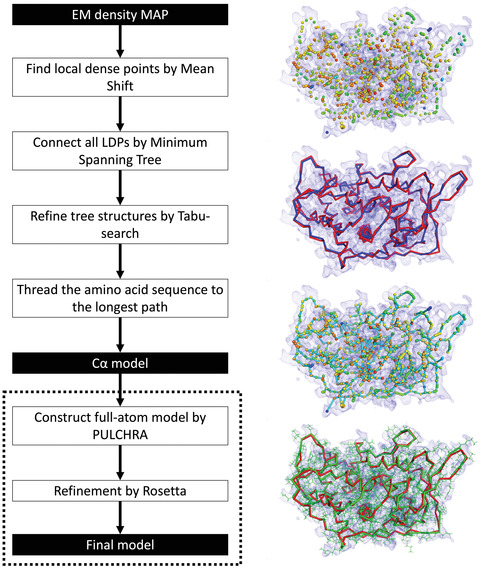
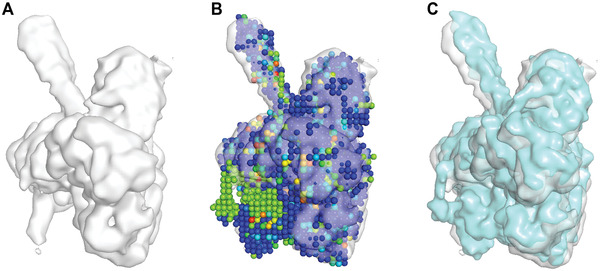
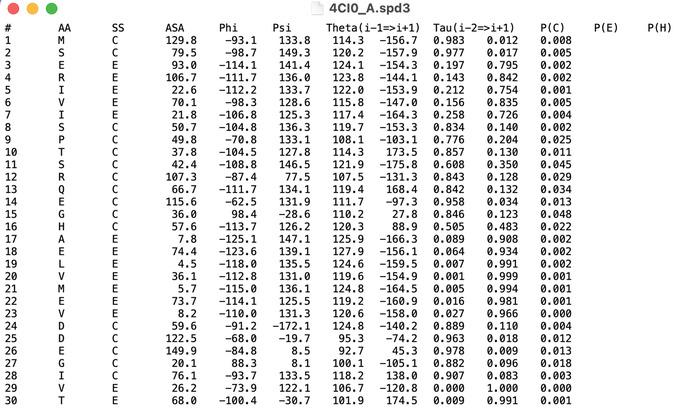
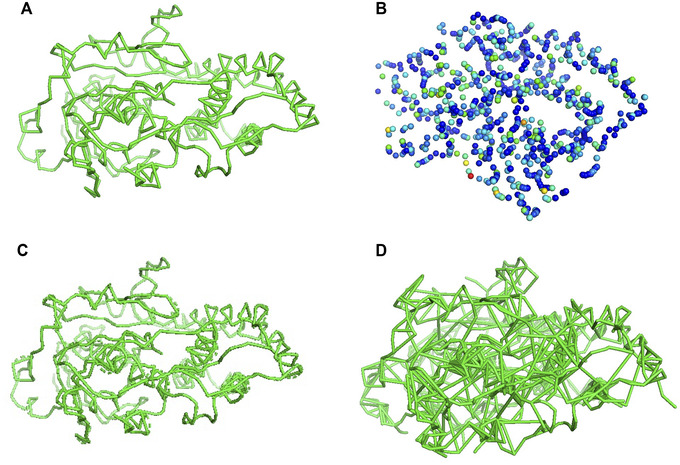
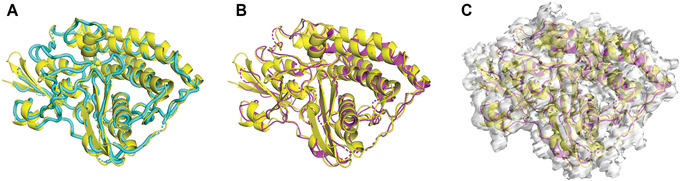
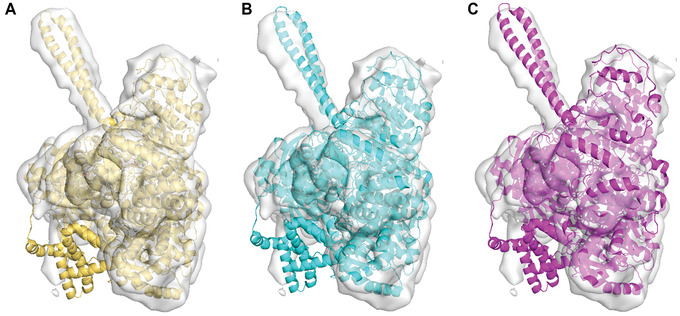
An increasing number of protein structures are determined by cryo-electron microscopy (cryo-EM) and stored in the Electron Microscopy Data Bank (EMDB). To interpret determined cryo-EM maps, several methods have been developed that model the tertiary structure of biomolecules, particularly proteins. Here we show how to use two such methods, VESPER and MAINMAST, which were developed in our group. VESPER is a method mainly for two purposes: fitting protein structure models into an EM map and aligning two EM maps locally or globally to capture their similarity. VESPER represents each EM map as a set of vectors pointing toward denser points. By considering matching the directions of vectors, in general, VESPER aligns maps better than conventional methods that only consider local densities of maps. MAINMAST is a de novo protein modeling tool designed for EM maps with resolution of 3-5 Å or better. MAINMAST builds a protein main chain directly from a density map by tracing dense points in an EM map and connecting them using a tree-graph structure. This article describes how to use these two tools using three illustrative modeling examples. © 2022 The Authors. Current Protocols published by Wiley Periodicals LLC. Basic Protocol 1: Protein structure model fitting using VESPER Alternate Protocol: Atomic model fitting using VESPER web server Basic Protocol 2: Protein de novo modeling using MAINMAST.
越来越多的蛋白质结构是通过冷冻电子显微镜(cryo-EM)确定并存储在电子显微镜数据银行(EMDB)中的。为了解释确定的冷冻电镜图谱,已经开发了几种方法来模拟生物分子的三级结构,特别是蛋白质的结构。在这里,我们展示如何使用我们小组开发的两种方法,VESPER 和 MAINMAST。VESPER 主要有两个目的:将蛋白质结构模型拟合到 EM 图谱中,以及局部或全局对齐两个 EM 图谱以捕捉它们的相似性。VESPER 将每个 EM 图谱表示为指向密集点的向量集。通过考虑匹配向量的方向,一般来说,VESPER 比仅考虑图谱局部密度的传统方法更好地对齐图谱。MAINMAST 是一种从头开始的蛋白质建模工具,专为分辨率为 3-5 Å 或更好的 EM 图谱设计。MAINMAST 通过在 EM 图谱中追踪密集点并用树图结构连接它们,直接从密度图谱构建蛋白质主链。本文描述了如何使用这两个工具,通过三个说明性建模示例进行说明。© 2022 作者。Wiley Periodicals LLC 出版的《当代协议》。基础方案 1:使用 VESPER 进行蛋白质结构模型拟合备选方案 1:使用 VESPER 网络服务器进行原子模型拟合基础方案 2:使用 MAINMAST 进行蛋白质从头建模。