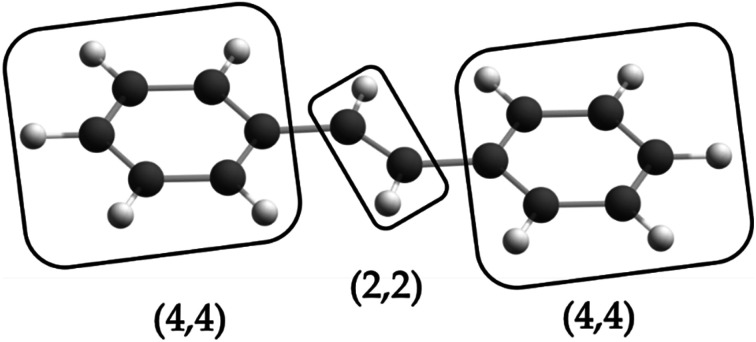
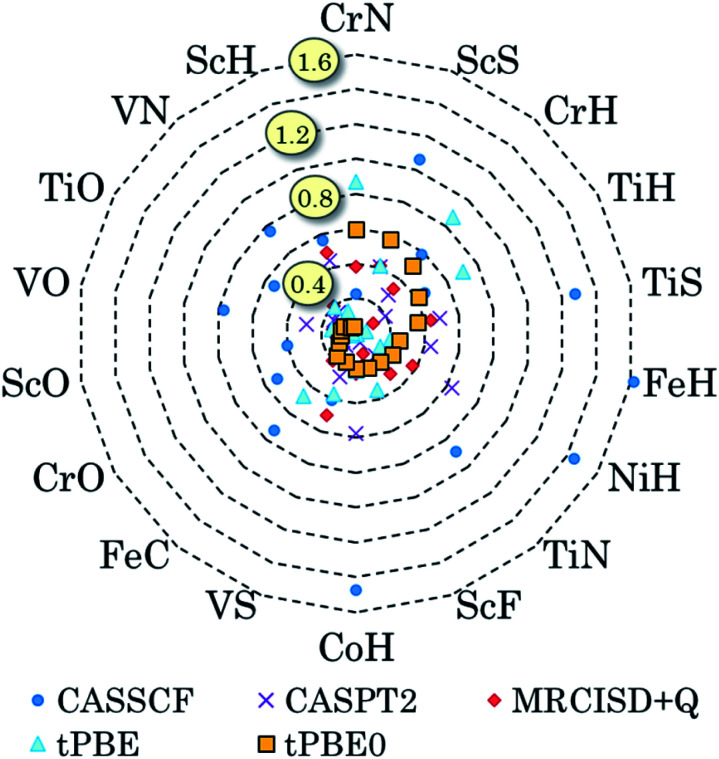
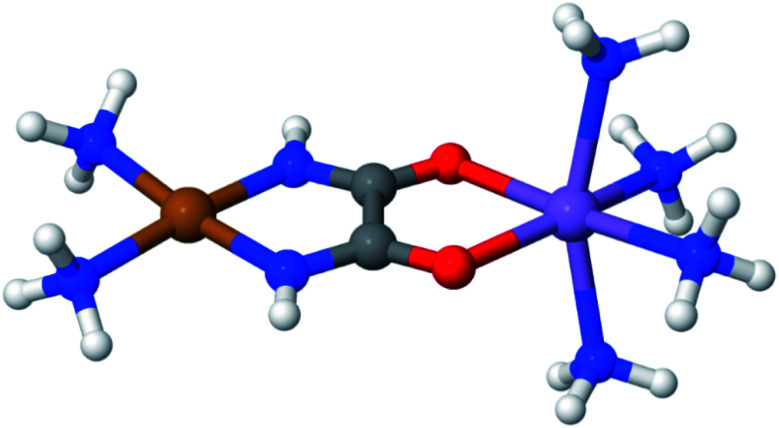
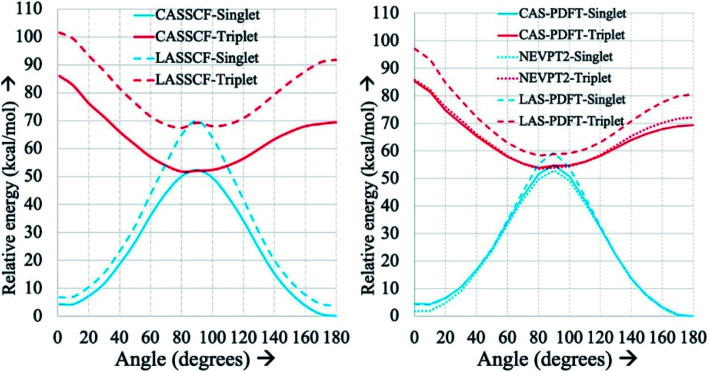
Zhou Chen, Hermes Matthew R, Wu Dihua, Bao Jie J, Pandharkar Riddhish, King Daniel S, Zhang Dayou, Scott Thais R, Lykhin Aleksandr O, Gagliardi Laura, Truhlar Donald G
Department of Chemistry, Chemical Theory Center, Minnesota Supercomputing Institute, University of Minnesota 207 Pleasant Street SE Minneapolis MN 55455-0431 USA
Department of Chemistry, Pritzker School of Molecular Engineering, The James Franck Institute and Chicago Center for Theoretical Chemistry, The University of Chicago Chicago IL 60637 USA
Chem Sci. 2022 Jun 7;13(26):7685-7706. doi: 10.1039/d2sc01022d. eCollection 2022 Jul 6.
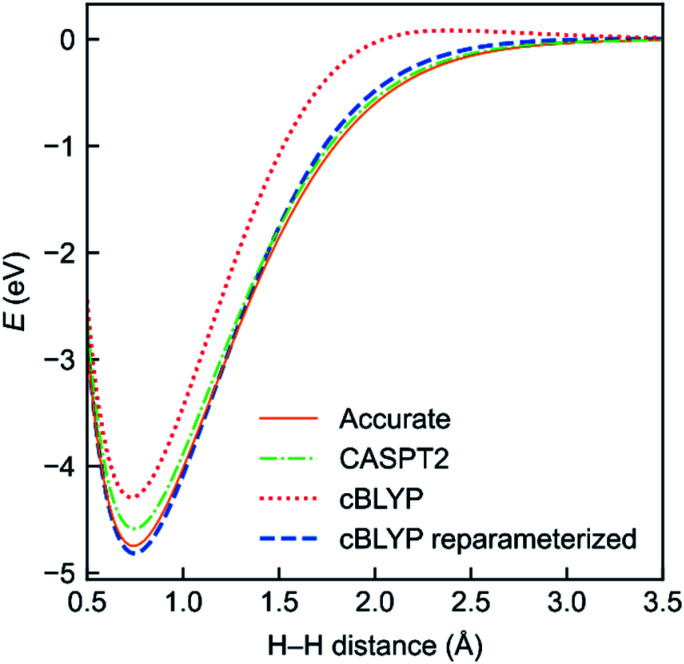
Strong electron correlation plays an important role in transition-metal and heavy-metal chemistry, magnetic molecules, bond breaking, biradicals, excited states, and many functional materials, but it provides a significant challenge for modern electronic structure theory. The treatment of strongly correlated systems usually requires a multireference method to adequately describe spin densities and near-degeneracy correlation. However, quantitative computation of dynamic correlation with multireference wave functions is often difficult or impractical. Multiconfiguration pair-density functional theory (MC-PDFT) provides a way to blend multiconfiguration wave function theory and density functional theory to quantitatively treat both near-degeneracy correlation and dynamic correlation in strongly correlated systems; it is more affordable than multireference perturbation theory, multireference configuration interaction, or multireference coupled cluster theory and more accurate for many properties than Kohn-Sham density functional theory. This perspective article provides a brief introduction to strongly correlated systems and previously reviewed progress on MC-PDFT followed by a discussion of several recent developments and applications of MC-PDFT and related methods, including localized-active-space MC-PDFT, generalized active-space MC-PDFT, density-matrix-renormalization-group MC-PDFT, hybrid MC-PDFT, multistate MC-PDFT, spin-orbit coupling, analytic gradients, and dipole moments. We also review the more recently introduced multiconfiguration nonclassical-energy functional theory (MC-NEFT), which is like MC-PDFT but allows for other ingredients in the nonclassical-energy functional. We discuss two new kinds of MC-NEFT methods, namely multiconfiguration density coherence functional theory and machine-learned functionals.
强电子关联在过渡金属和重金属化学、磁性分子、键断裂、双自由基、激发态以及许多功能材料中起着重要作用,但它给现代电子结构理论带来了重大挑战。处理强关联体系通常需要多参考方法来充分描述自旋密度和近简并关联。然而,用多参考波函数对动态关联进行定量计算往往困难或不切实际。多组态对密度泛函理论(MC-PDFT)提供了一种将多组态波函数理论和密度泛函理论相结合的方法,以定量处理强关联体系中的近简并关联和动态关联;它比多参考微扰理论、多参考组态相互作用或多参考耦合簇理论成本更低,并且对于许多性质而言比科恩-沈密度泛函理论更准确。这篇观点文章简要介绍了强关联体系以及之前关于MC-PDFT的综述进展,随后讨论了MC-PDFT及相关方法的一些最新发展和应用,包括局域活性空间MC-PDFT、广义活性空间MC-PDFT、密度矩阵重整化群MC-PDFT、混合MC-PDFT、多态MC-PDFT、自旋-轨道耦合、解析梯度和偶极矩。我们还综述了最近引入的多组态非经典能量泛函理论(MC-NEFT),它与MC-PDFT类似,但在非经典能量泛函中允许有其他成分。我们讨论了两种新型的MC-NEFT方法,即多组态密度相干泛函理论和机器学习泛函。