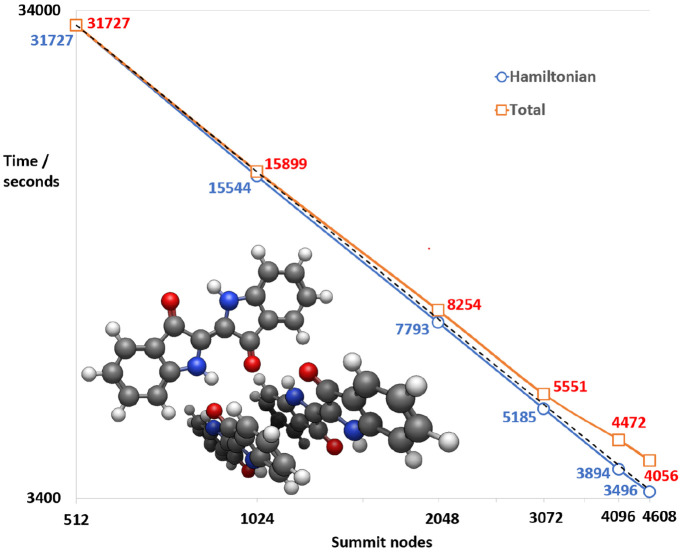
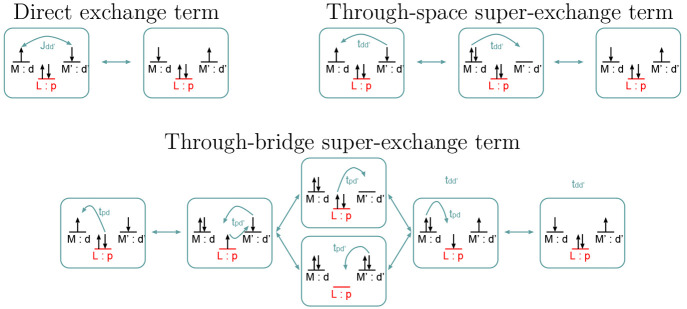
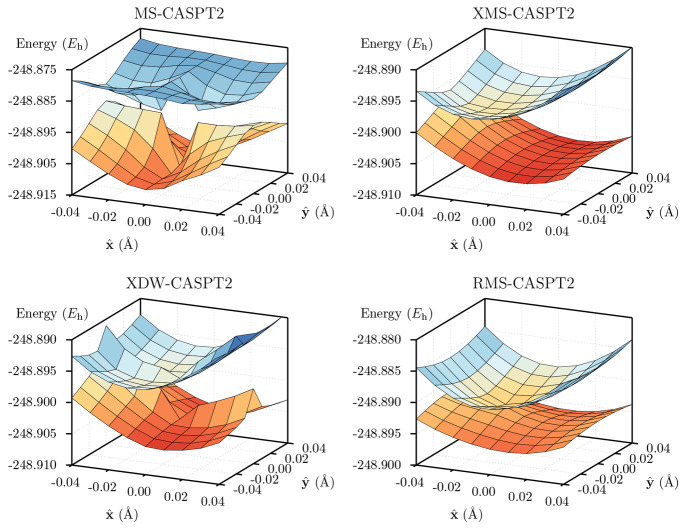
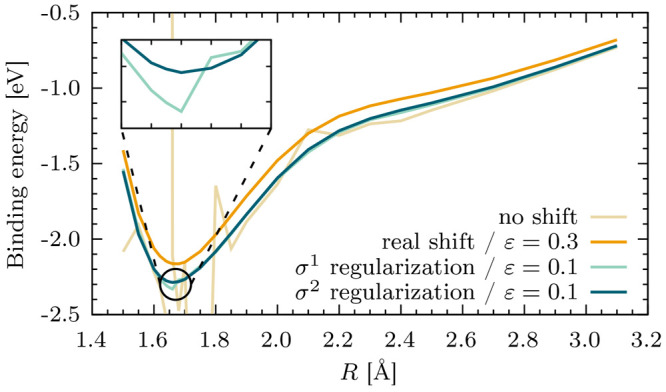
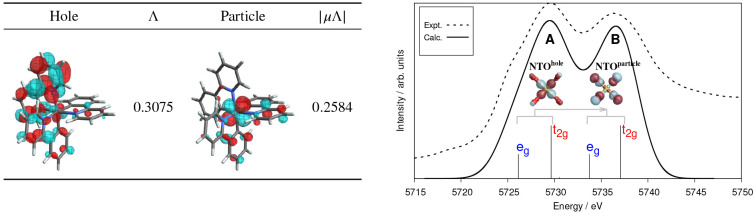
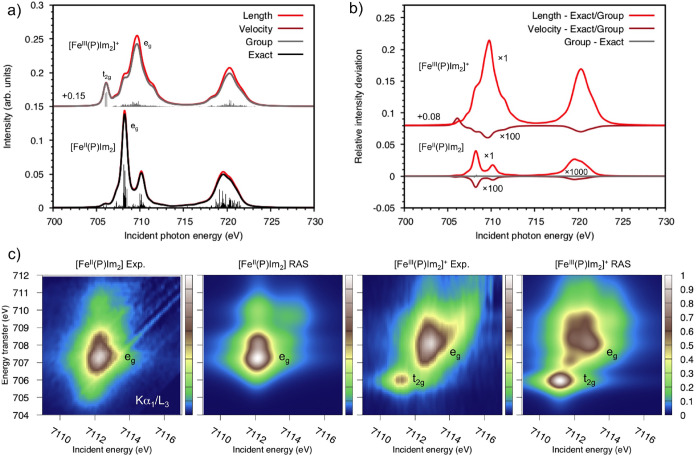
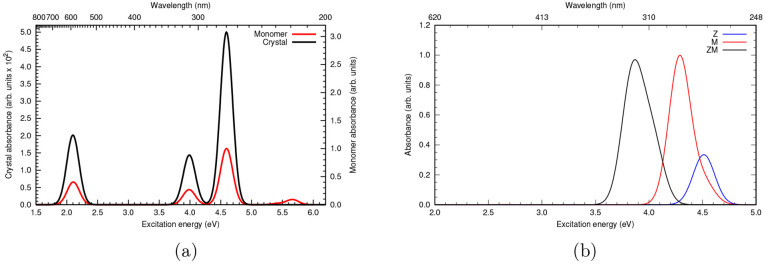
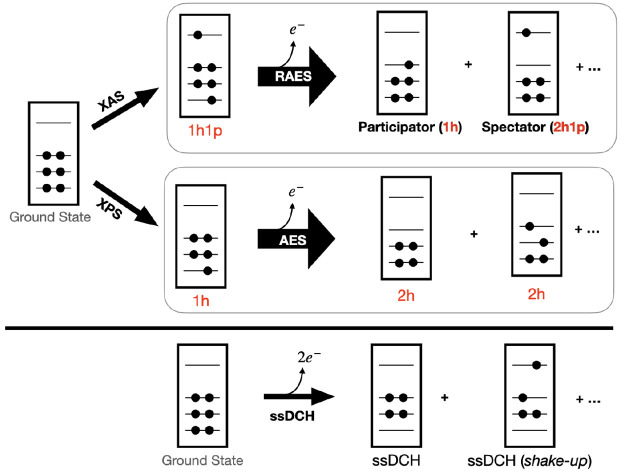
Li Manni Giovanni, Fdez Galván Ignacio, Alavi Ali, Aleotti Flavia, Aquilante Francesco, Autschbach Jochen, Avagliano Davide, Baiardi Alberto, Bao Jie J, Battaglia Stefano, Birnoschi Letitia, Blanco-González Alejandro, Bokarev Sergey I, Broer Ria, Cacciari Roberto, Calio Paul B, Carlson Rebecca K, Carvalho Couto Rafael, Cerdán Luis, Chibotaru Liviu F, Chilton Nicholas F, Church Jonathan Richard, Conti Irene, Coriani Sonia, Cuéllar-Zuquin Juliana, Daoud Razan E, Dattani Nike, Decleva Piero, de Graaf Coen, Delcey Mickaël G, De Vico Luca, Dobrautz Werner, Dong Sijia S, Feng Rulin, Ferré Nicolas, Filatov Gulak Michael, Gagliardi Laura, Garavelli Marco, González Leticia, Guan Yafu, Guo Meiyuan, Hennefarth Matthew R, Hermes Matthew R, Hoyer Chad E, Huix-Rotllant Miquel, Jaiswal Vishal Kumar, Kaiser Andy, Kaliakin Danil S, Khamesian Marjan, King Daniel S, Kochetov Vladislav, Krośnicki Marek, Kumaar Arpit Arun, Larsson Ernst D, Lehtola Susi, Lepetit Marie-Bernadette, Lischka Hans, López Ríos Pablo, Lundberg Marcus, Ma Dongxia, Mai Sebastian, Marquetand Philipp, Merritt Isabella C D, Montorsi Francesco, Mörchen Maximilian, Nenov Artur, Nguyen Vu Ha Anh, Nishimoto Yoshio, Oakley Meagan S, Olivucci Massimo, Oppel Markus, Padula Daniele, Pandharkar Riddhish, Phung Quan Manh, Plasser Felix, Raggi Gerardo, Rebolini Elisa, Reiher Markus, Rivalta Ivan, Roca-Sanjuán Daniel, Romig Thies, Safari Arta Anushirwan, Sánchez-Mansilla Aitor, Sand Andrew M, Schapiro Igor, Scott Thais R, Segarra-Martí Javier, Segatta Francesco, Sergentu Dumitru-Claudiu, Sharma Prachi, Shepard Ron, Shu Yinan, Staab Jakob K, Straatsma Tjerk P, Sørensen Lasse Kragh, Tenorio Bruno Nunes Cabral, Truhlar Donald G, Ungur Liviu, Vacher Morgane, Veryazov Valera, Voß Torben Arne, Weser Oskar, Wu Dihua, Yang Xuchun, Yarkony David, Zhou Chen, Zobel J Patrick, Lindh Roland
Electronic Structure Theory Department, Max Planck Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany.
Department of Chemistry - BMC, Uppsala University, P.O. Box 576, SE-75123 Uppsala, Sweden.
J Chem Theory Comput. 2023 Oct 24;19(20):6933-6991. doi: 10.1021/acs.jctc.3c00182. Epub 2023 May 22.
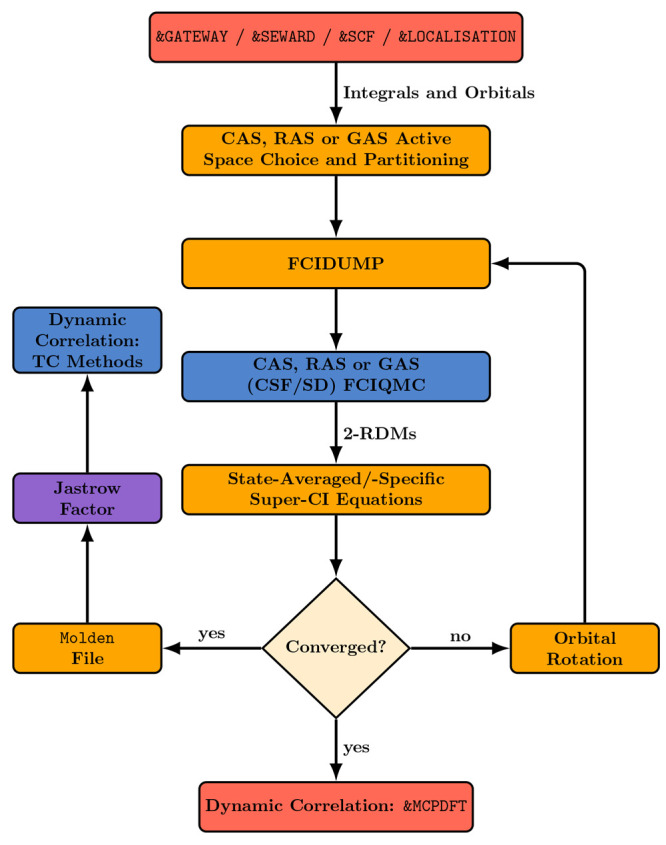
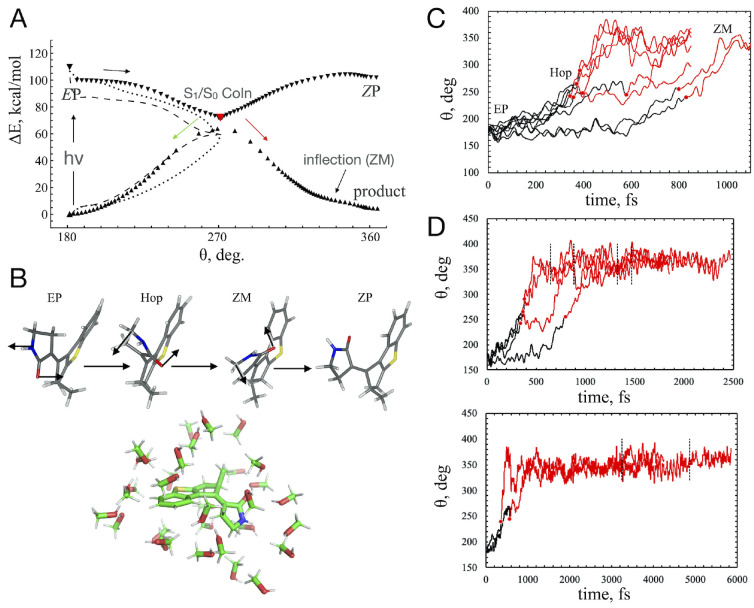
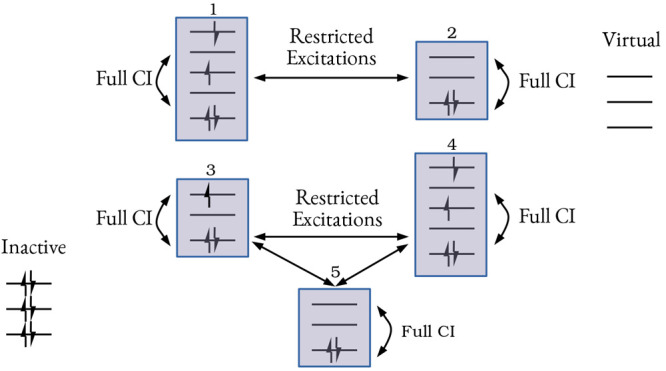
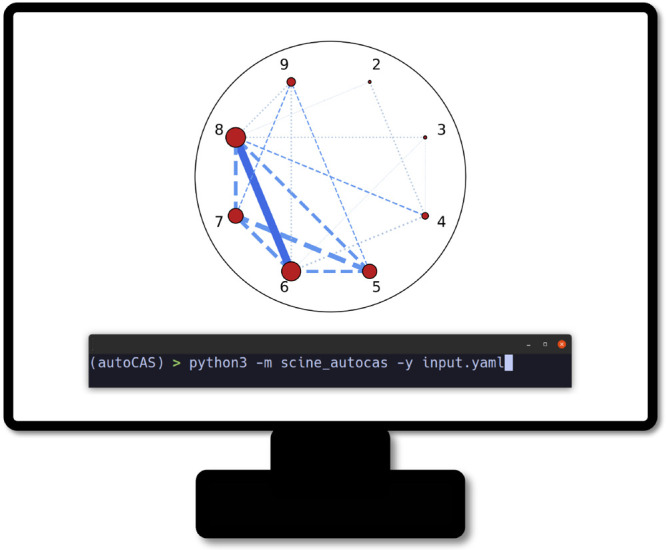
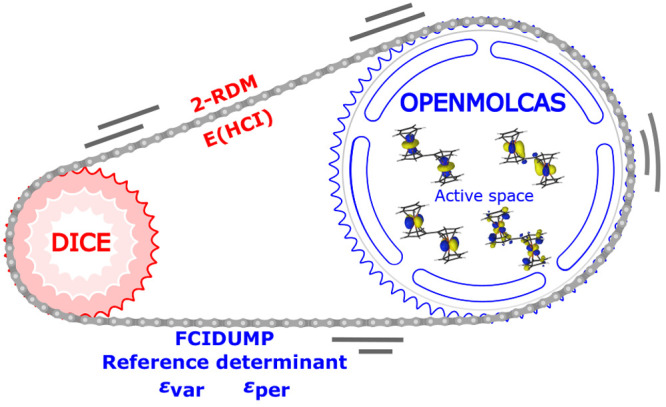
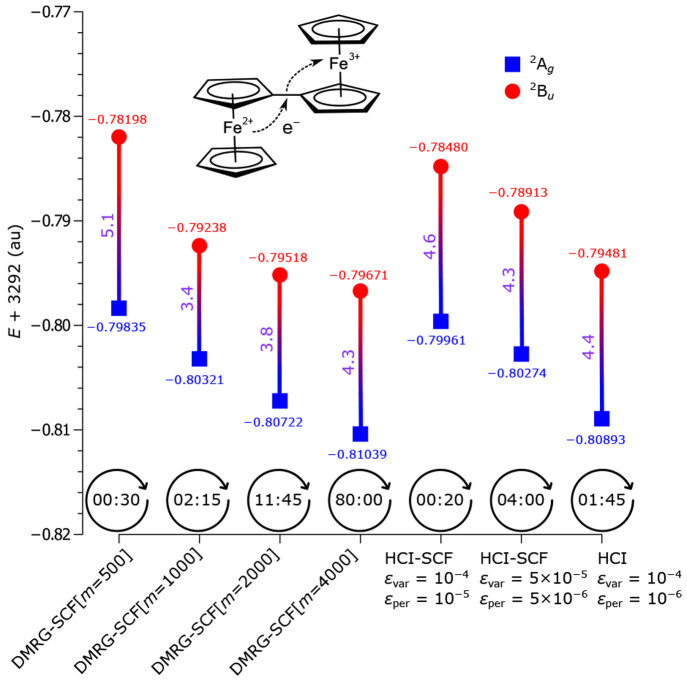
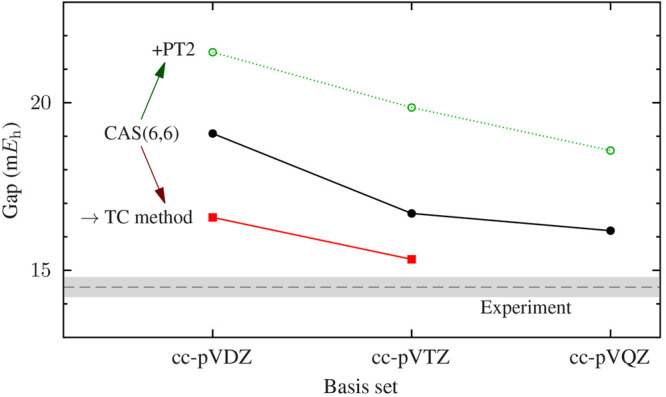
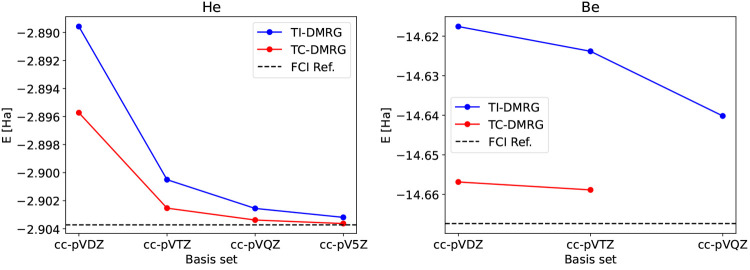
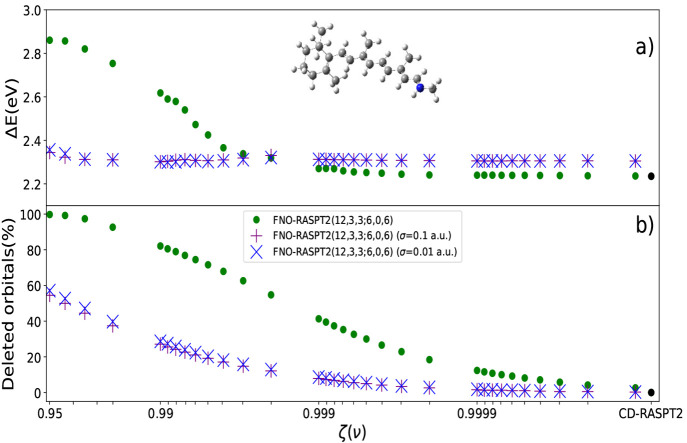
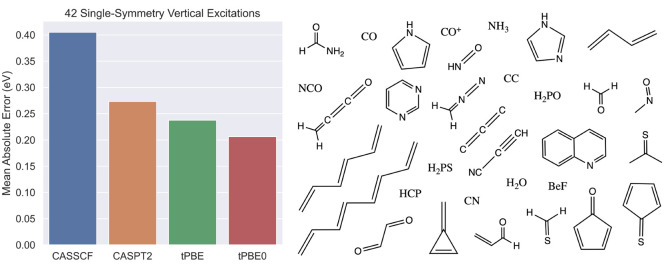
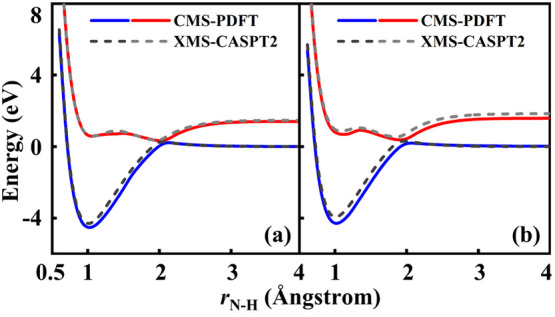
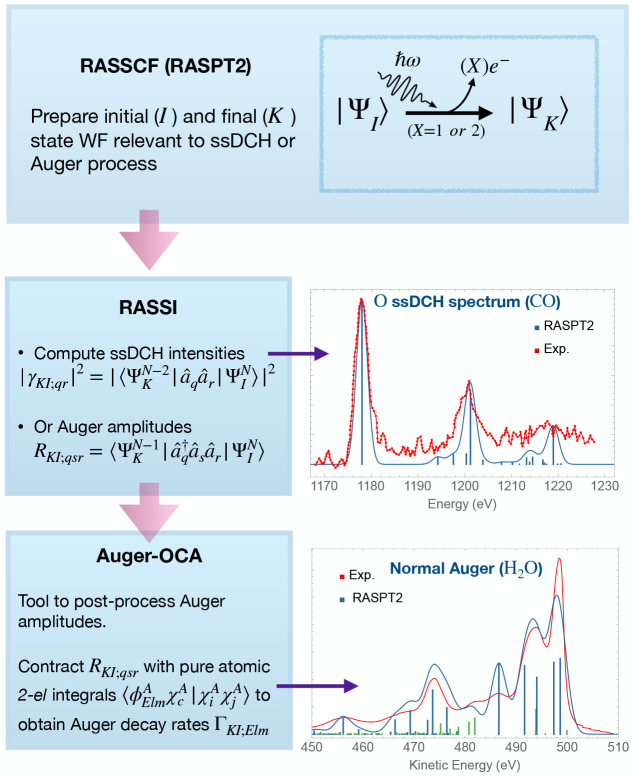
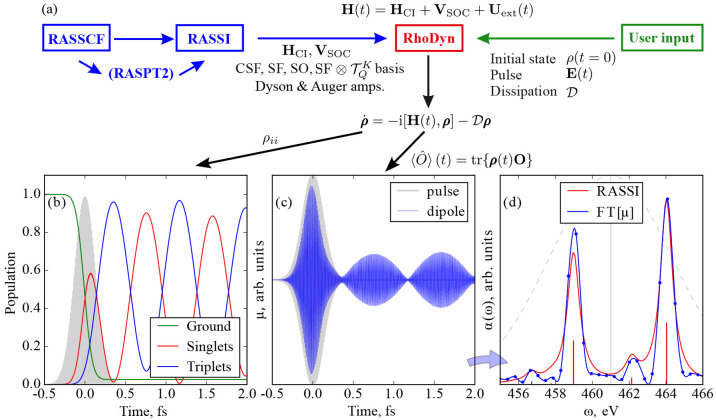
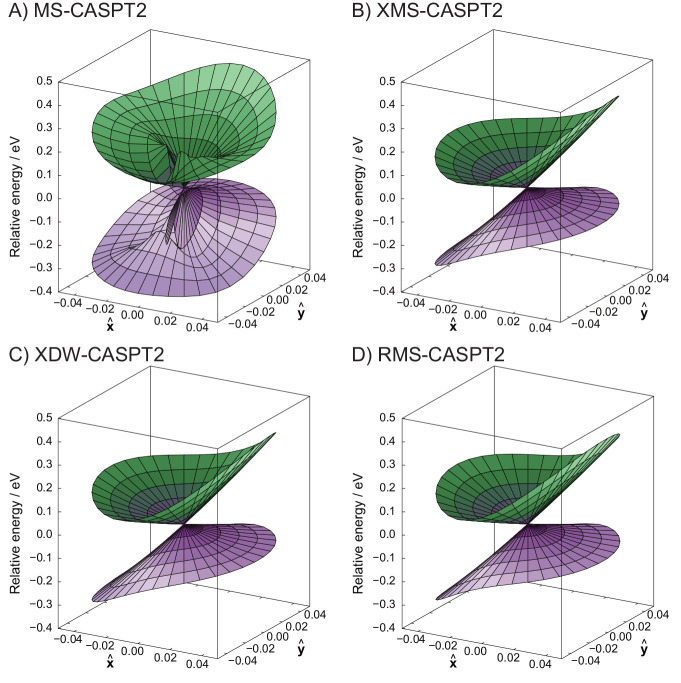
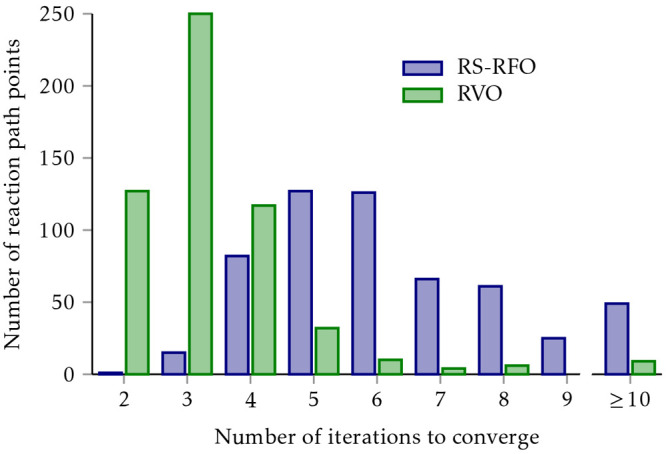
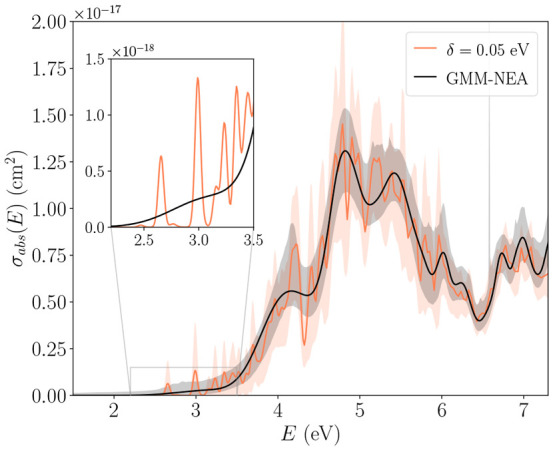
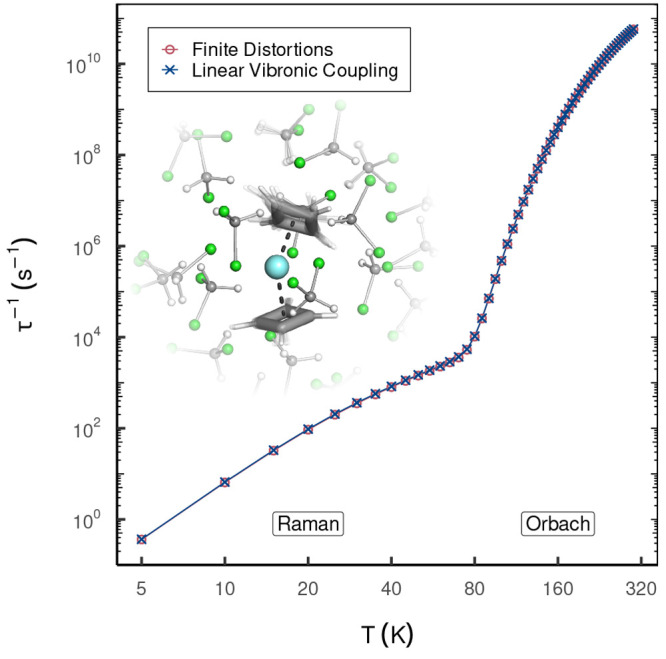
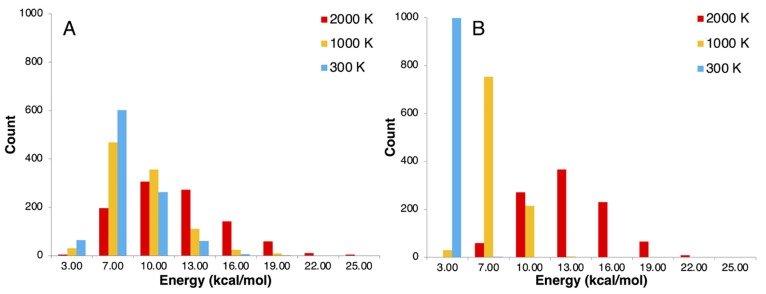
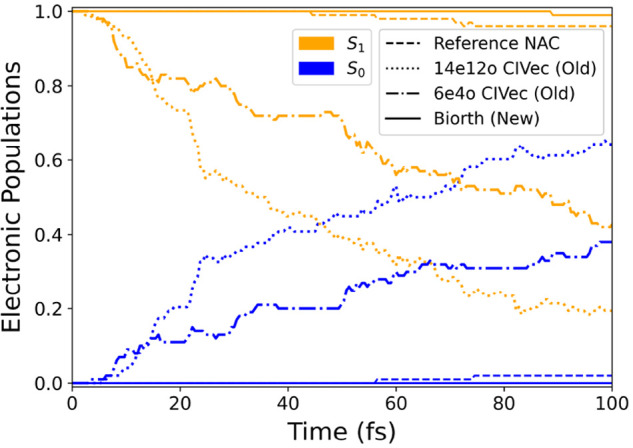
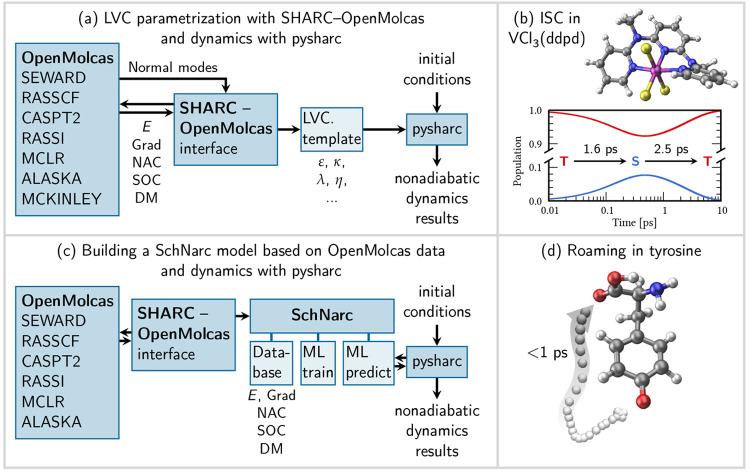
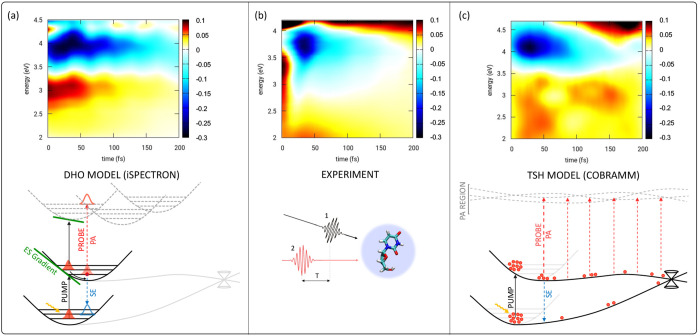
The developments of the open-source OpenMolcas chemistry software environment since spring 2020 are described, with a focus on novel functionalities accessible in the stable branch of the package or via interfaces with other packages. These developments span a wide range of topics in computational chemistry and are presented in thematic sections: electronic structure theory, electronic spectroscopy simulations, analytic gradients and molecular structure optimizations, ab initio molecular dynamics, and other new features. This report offers an overview of the chemical phenomena and processes OpenMolcas can address, while showing that OpenMolcas is an attractive platform for state-of-the-art atomistic computer simulations.
本文描述了开源的OpenMolcas化学软件环境自2020年春季以来的发展情况,重点介绍了该软件包稳定分支中可获取的新功能或通过与其他软件包的接口实现的新功能。这些发展涵盖了计算化学中的广泛主题,并在主题部分进行了介绍:电子结构理论、电子光谱模拟、解析梯度和分子结构优化、从头算分子动力学以及其他新特性。本报告概述了OpenMolcas可以处理的化学现象和过程,同时表明OpenMolcas是进行前沿原子级计算机模拟的一个有吸引力的平台。