The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui, China.
School of Life Sciences, University of Science and Technology of China, Hefei, Anhui, China.
BMC Microbiol. 2022 Jul 23;22(1):184. doi: 10.1186/s12866-022-02594-y.
Gastric microbial dysbiosis were reported to be associated with gastric cancer (GC). This study aimed to explore the variation, diversity, and composition patterns of gastric bacteria in stages of gastric carcinogenesis based on the published datasets.
We conducted a gastric microbial analysis using 10 public datasets based on 16S rRNA sequencing, including 1270 gastric biopsies of 109 health control, 183 superficial gastritis (SG), 135 atrophic gastritis (AG), 124 intestinal metaplasia (IM), 94 intraepithelial neoplasia (IN), 344 GC, and 281 adjacent normal tissues. And QIIME2-pipeline, DESeq2, NetMoss2, vegan, igraph, and RandomForest were used for the data processing and analysis.
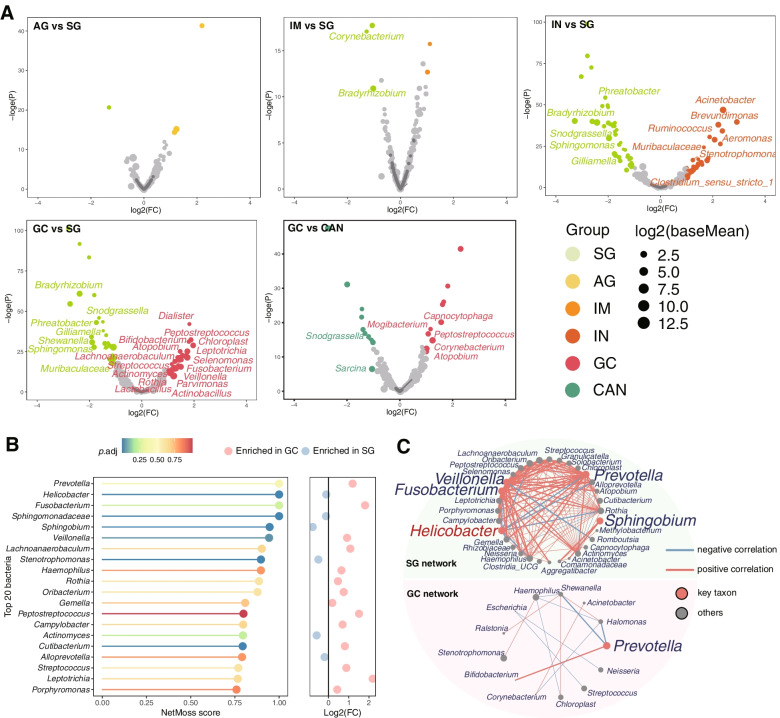
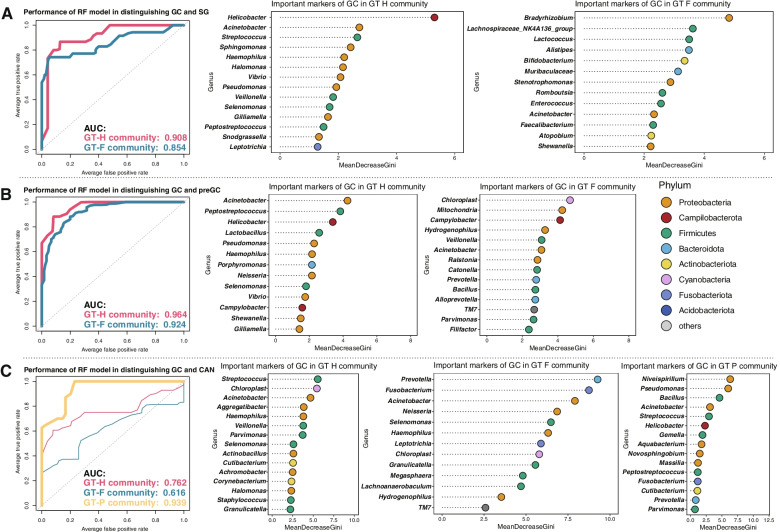
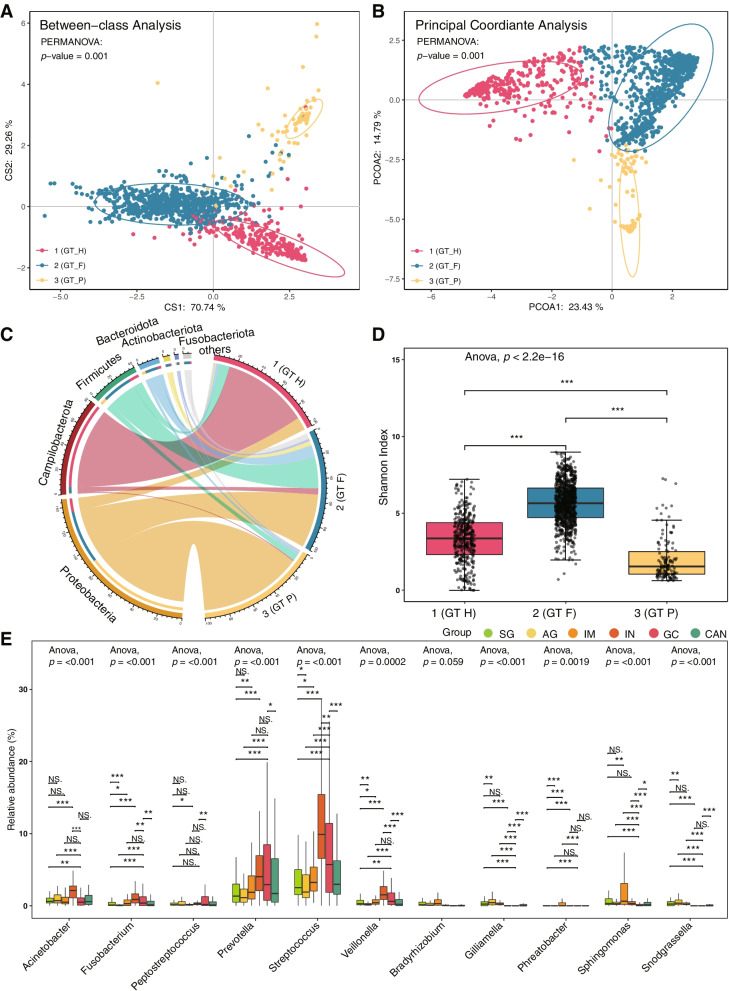
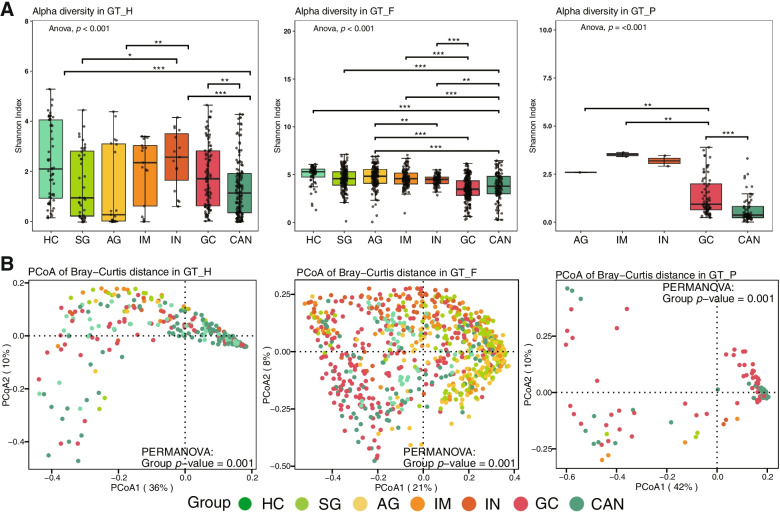
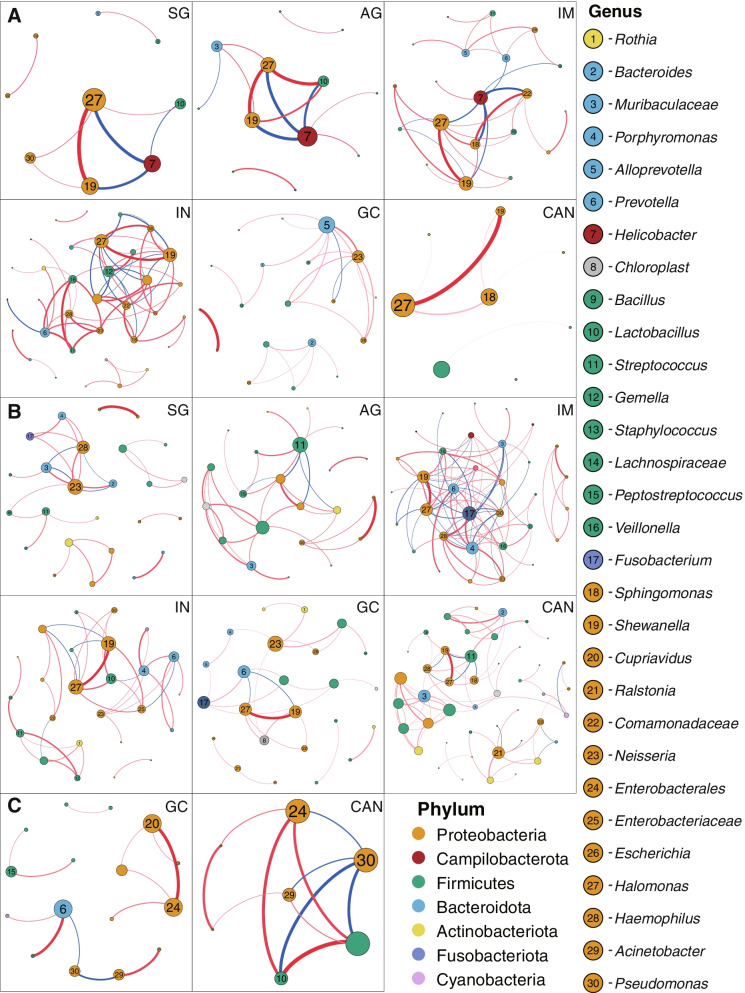
We identified three gastric microbial communities among all the gastric tissues. The first community (designate as GT-H) was featured by the high abundance of Helicobacter. The other two microbial communities, namely GT-F, and GT-P, were featured by the enrichment of phylum Firmicutes and Proteobacteria, respectively. The distribution of GC-associated bacteria, such as Fusobacterium, Peptostreptococcus, Streptococcus, and Veillonella were enriched in tumor tissues, and mainly distributed in GT-F type microbial communities. Compared with SG, AG, and IM, the bacterial diversity in GC was significantly reduced. And the strength of microbial interaction networks was initially increased in IM but gradually decreased from IN to GC. In addition, Randomforest models constructed in in GT-H and GT-F microbial communities showed excellent performance in distinguishing GC from SG and precancerous stages, with varied donated bacteria.
This study identified three types of gastric microbiome with different patterns of composition which helps to clarify the potential key bacteria in the development of gastric carcinogenesis.
胃微生物失调与胃癌(GC)有关。本研究旨在基于已发表的数据集,探索胃癌发生过程中胃细菌的变化、多样性和组成模式。
我们使用基于 16S rRNA 测序的 10 个公共数据集进行了胃微生物分析,包括 109 例健康对照、183 例浅表性胃炎(SG)、135 例萎缩性胃炎(AG)、124 例肠上皮化生(IM)、94 例上皮内瘤变(IN)、344 例 GC 和 281 例相邻正常组织的 1270 个胃活检。并使用 QIIME2 管道、DESeq2、NetMoss2、vegan、igraph 和 RandomForest 进行数据处理和分析。
我们在所有胃组织中鉴定出三个胃微生物群落。第一个群落(指定为 GT-H)的特征是幽门螺杆菌的高丰度。其他两个微生物群落,即 GT-F 和 GT-P,分别以厚壁菌门和变形菌门的丰度富集为特征。GC 相关细菌(如梭杆菌属、消化链球菌属、链球菌属和韦荣球菌属)的分布在肿瘤组织中富集,主要分布在 GT-F 型微生物群落中。与 SG、AG 和 IM 相比,GC 中的细菌多样性显著降低。并且从 IN 到 GC,微生物相互作用网络的强度最初增加,然后逐渐降低。此外,在 GT-H 和 GT-F 微生物群落中构建的 Randomforest 模型在区分 GC 与 SG 和癌前阶段方面表现出优异的性能,具有不同的捐赠细菌。
本研究鉴定了三种具有不同组成模式的胃微生物群,有助于阐明胃癌发生过程中潜在的关键细菌。