Medical Research Council Functional Genomics Unit, Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, United Kingdom.
School of Life Science and Technology, Shanghai, Tech University, Shanghai, China.
PLoS Genet. 2022 Jul 25;18(7):e1010325. doi: 10.1371/journal.pgen.1010325. eCollection 2022 Jul.
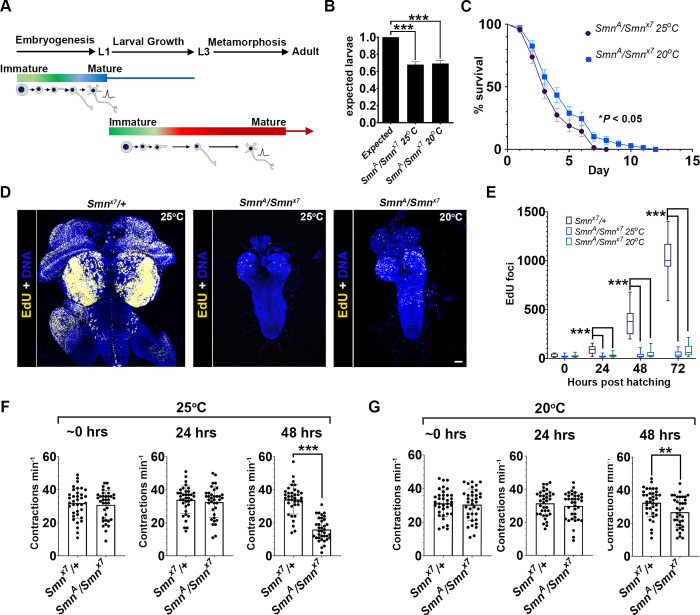
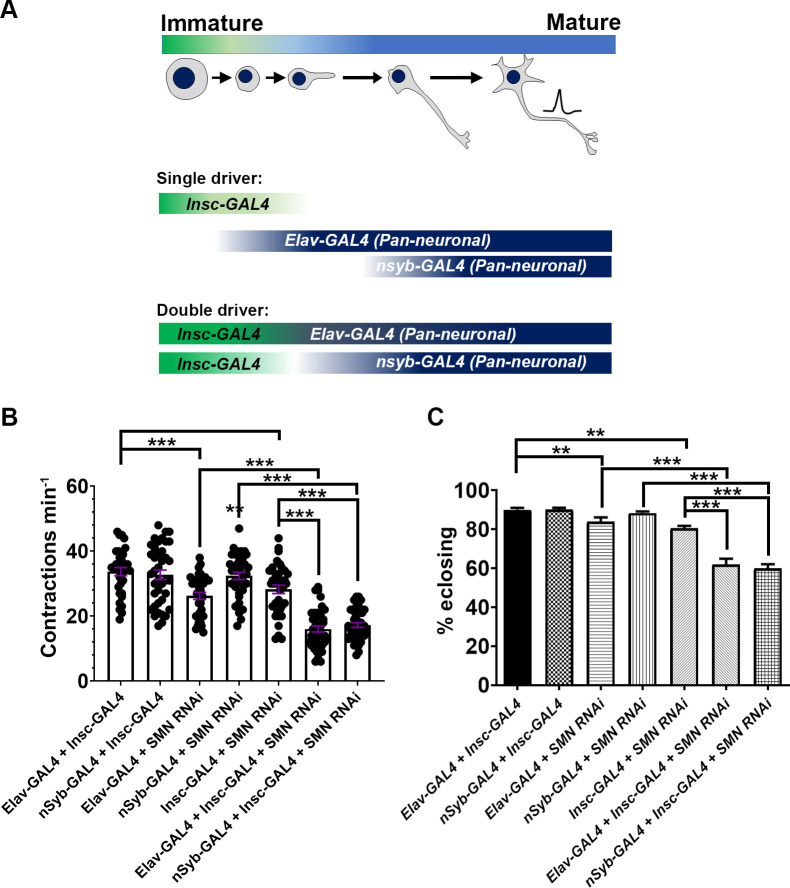
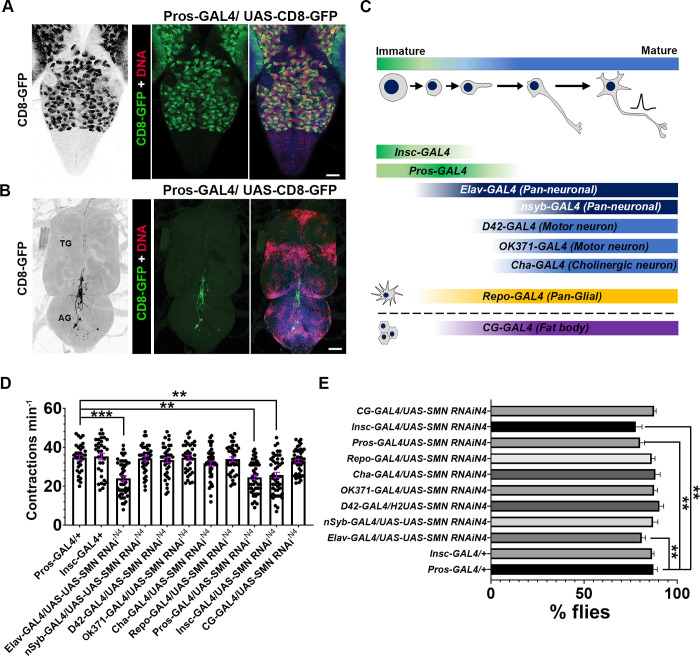
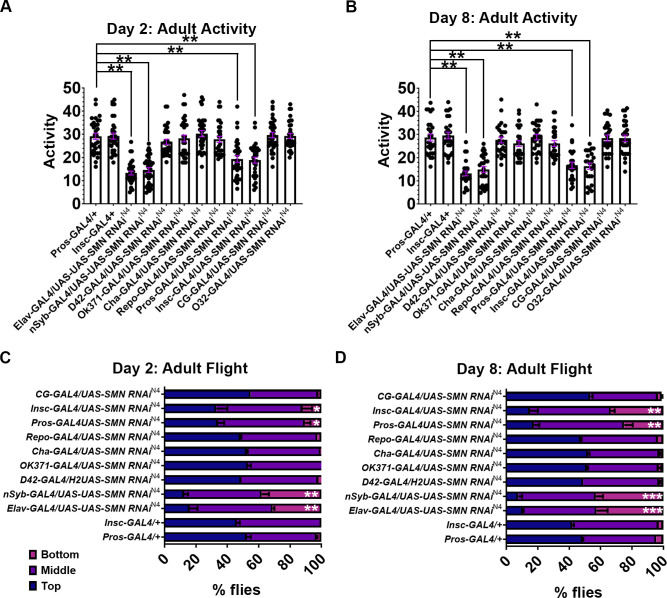
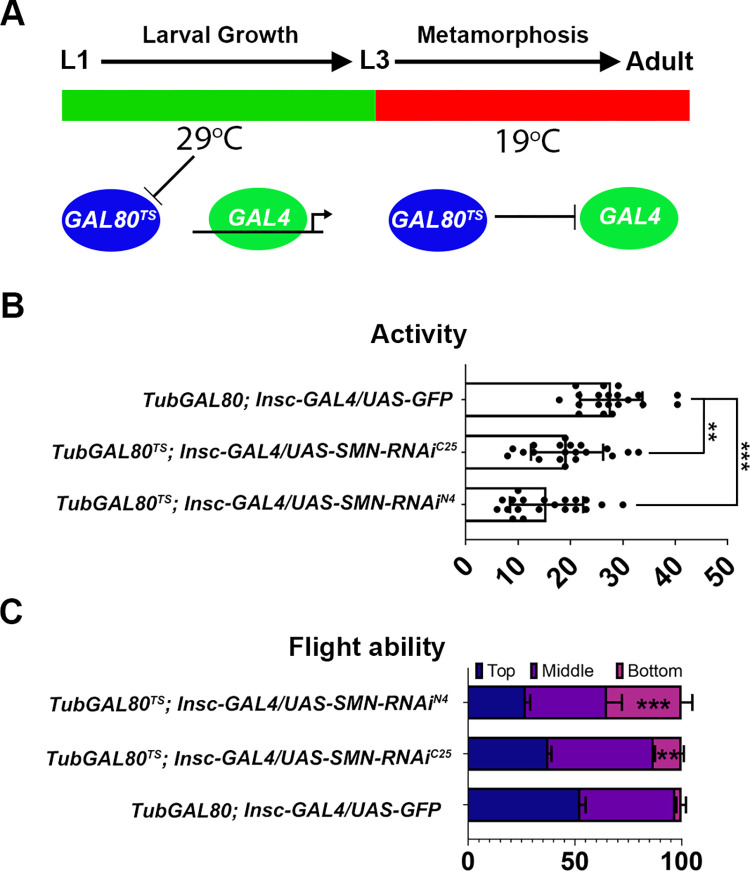
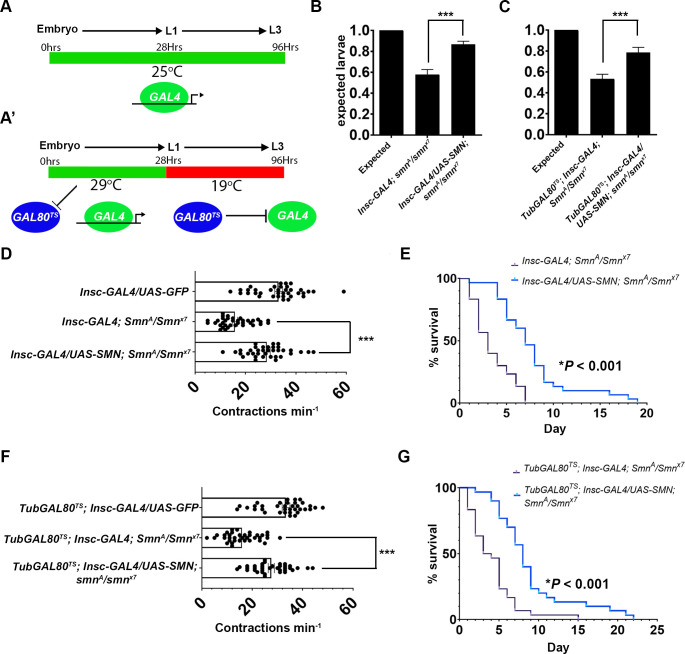
Spinal muscular atrophy (SMA) is the most common autosomal recessive neurodegenerative disease, and is characterised by spinal motor neuron loss, impaired motor function and, often, premature death. Mutations and deletions in the widely expressed survival motor neuron 1 (SMN1) gene cause SMA; however, the mechanisms underlying the selectivity of motor neuron degeneration are not well understood. Although SMA is degenerative in nature, SMN function during embryonic and early postnatal development appears to be essential for motor neuron survival in animal models and humans. Notwithstanding, how developmental defects contribute to the subversion of postnatal and adult motor function remains elusive. Here, in a Drosophila SMA model, we show that neurodevelopmental defects precede gross locomotor dysfunction in larvae. Furthermore, to specifically address the relevance of SMN during neurogenesis and in neurogenic cell types, we show that SMN knockdown using neuroblast-specific and pan-neuronal drivers, but not differentiated neuron or glial cell drivers, impairs adult motor function. Using targeted knockdown, we further restricted SMN manipulation in neuroblasts to a defined time window. Our aim was to express specifically in the neuronal progenitor cell types that have not formed synapses, and thus a time that precedes neuromuscular junction formation and maturation. By restoring SMN levels in these distinct neuronal population, we partially rescue the larval locomotor defects of Smn mutants. Finally, combinatorial SMN knockdown in immature and mature neurons synergistically enhances the locomotor and survival phenotypes. Our in-vivo study is the first to directly rescue the motor defects of an SMA model by expressing Smn in an identifiable population of Drosophila neuroblasts and developing neurons, highlighting that neuronal sensitivity to SMN loss may arise before synapse establishment and nerve cell maturation.
脊髓性肌萎缩症(SMA)是最常见的常染色体隐性神经退行性疾病,其特征是脊髓运动神经元丧失、运动功能受损,并且通常导致早逝。广泛表达的运动神经元生存 1 号(SMN1)基因的突变和缺失导致 SMA;然而,运动神经元退化的选择性机制尚未得到很好的理解。尽管 SMA 具有退行性,但在动物模型和人类中,SMN 在胚胎和早期出生后发育过程中的功能似乎对运动神经元的存活至关重要。尽管如此,发育缺陷如何导致出生后和成年运动功能的破坏仍然难以捉摸。在这里,在果蝇 SMA 模型中,我们表明神经发育缺陷先于幼虫的运动功能障碍。此外,为了专门研究 SMN 在神经发生和神经发生细胞类型中的相关性,我们表明,使用神经母细胞特异性和全神经元驱动子而非分化神经元或神经胶质细胞驱动子进行 SMN 敲低会损害成年运动功能。使用靶向敲低,我们进一步将 SMN 操作限制在神经母细胞中的特定时间窗口。我们的目的是在尚未形成突触的神经祖细胞类型中特异性表达 SMN,从而在形成和成熟神经肌肉接头之前。通过在这些不同的神经元群体中特异性恢复 SMN 水平,我们部分挽救了 Smn 突变体的幼虫运动缺陷。最后,不成熟和成熟神经元中的组合 SMN 敲低协同增强了运动和存活表型。我们的体内研究是第一个通过在果蝇神经母细胞和发育神经元中可识别的群体中表达 Smn 来直接挽救 SMA 模型的运动缺陷的研究,突出了神经元对 SMN 丢失的敏感性可能在突触建立和神经细胞成熟之前出现。