Department of Chemistry, Yeshiva College, Yeshiva University, New York, NY, 10033, USA.
Department of Systems and Computational Biology, Albert Einstein College of Medicine, Bronx, NY, 10461, USA.
BMC Bioinformatics. 2022 Jul 25;23(1):301. doi: 10.1186/s12859-022-04852-2.
Identifying protein interfaces can inform how proteins interact with their binding partners, uncover the regulatory mechanisms that control biological functions and guide the development of novel therapeutic agents. A variety of computational approaches have been developed for predicting a protein's interfacial residues from its known sequence and structure. Methods using the known three-dimensional structures of proteins can be template-based or template-free. Template-based methods have limited success in predicting interfaces when homologues with known complex structures are not available to use as templates. The prediction performance of template-free methods that only rely only upon proteins' intrinsic properties is limited by the amount of biologically relevant features that can be included in an interface prediction model.
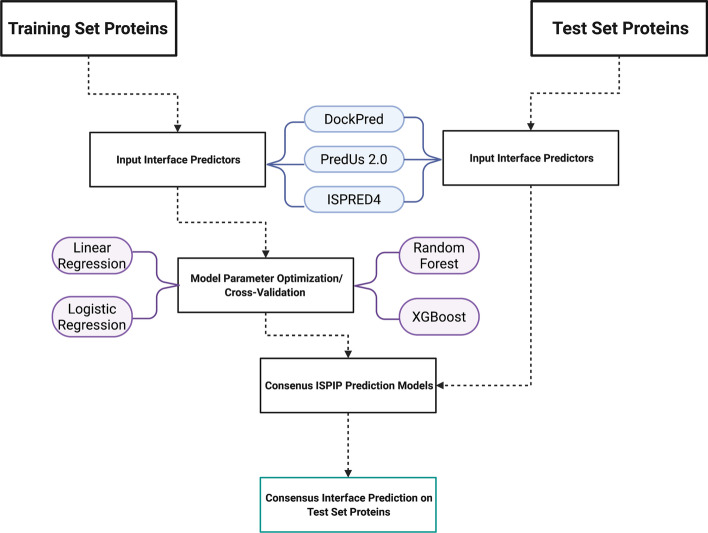
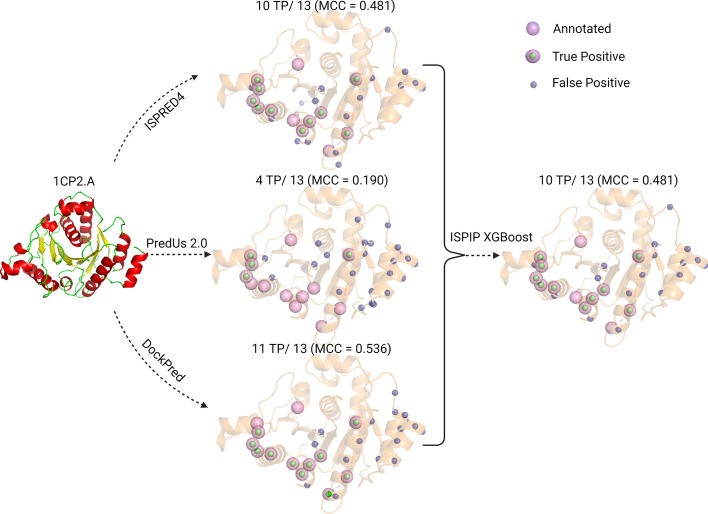
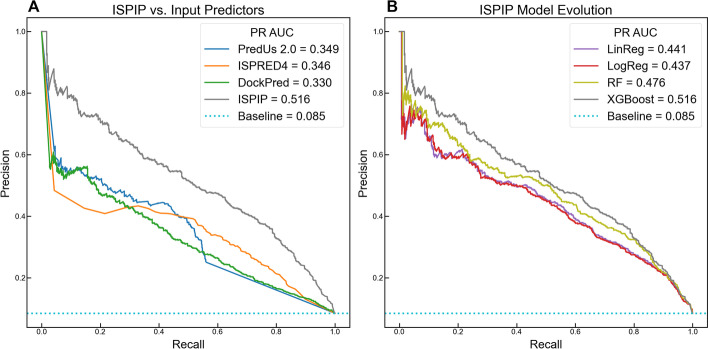
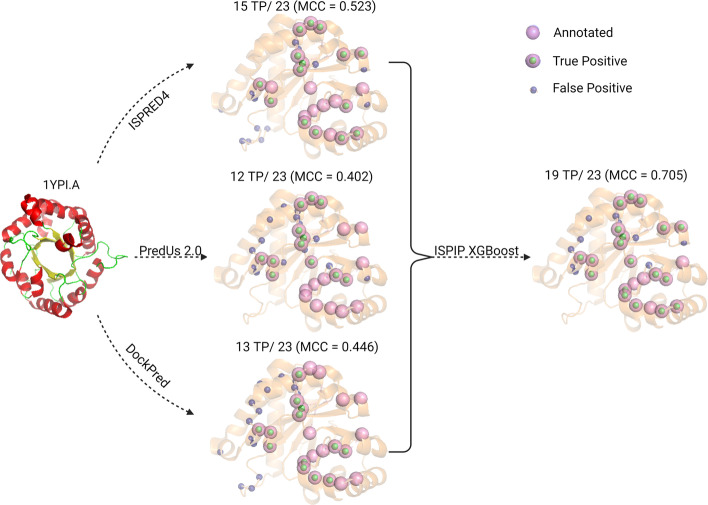
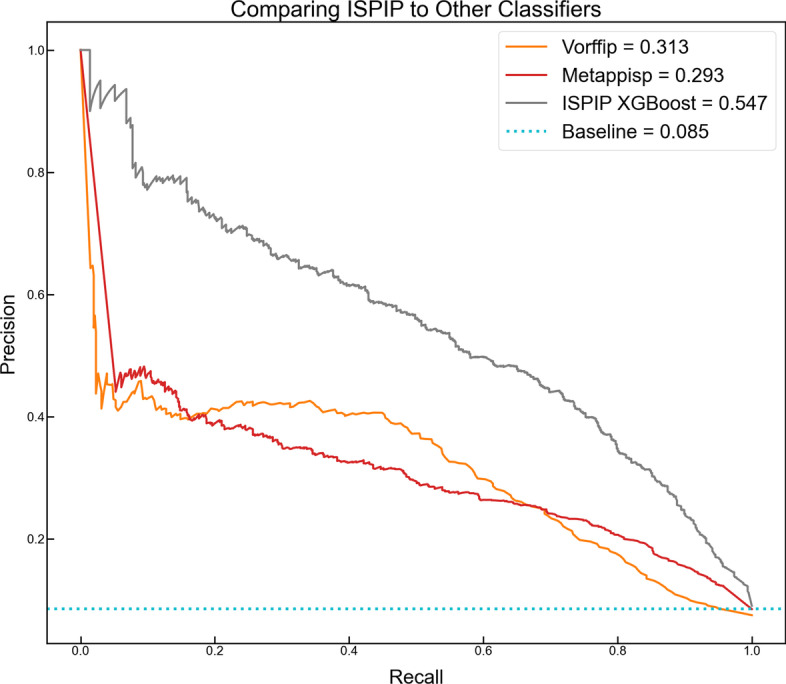
We describe the development of an integrated method for protein interface prediction (ISPIP) to explore the hypothesis that the efficacy of a computational prediction method of protein binding sites can be enhanced by using a combination of methods that rely on orthogonal structure-based properties of a query protein, combining and balancing both template-free and template-based features. ISPIP is a method that integrates these approaches through simple linear or logistic regression models and more complex decision tree models. On a diverse test set of 156 query proteins, ISPIP outperforms each of its individual classifiers in identifying protein binding interfaces.
The integrated method captures the best performance of individual classifiers and delivers an improved interface prediction. The method is robust and performs well even when one of the individual classifiers performs poorly on a particular query protein. This work demonstrates that integrating orthogonal methods that depend on different structural properties of proteins performs better at interface prediction than any individual classifier alone.
鉴定蛋白质界面可以揭示蛋白质与其结合伴侣相互作用的方式,揭示控制生物功能的调控机制,并指导新型治疗剂的开发。已经开发了多种从已知序列和结构预测蛋白质界面残基的计算方法。使用蛋白质已知三维结构的方法可以是基于模板的或无模板的。当没有已知复合物结构的同源物可用作模板时,基于模板的方法在预测界面方面的成功率有限。仅依赖蛋白质固有特性的无模板方法的预测性能受到可以包含在接口预测模型中的生物学相关特征的数量的限制。
我们描述了一种用于蛋白质界面预测(ISPIP)的综合方法的开发,以探索以下假设:通过使用组合方法来增强计算蛋白质结合位点预测方法的功效,这些方法依赖于查询蛋白质的正交基于结构的特性,结合和平衡无模板和基于模板的特征。ISPIP 通过简单的线性或逻辑回归模型以及更复杂的决策树模型来集成这些方法。在包含 156 个查询蛋白的多样化测试集中,ISPIP 在识别蛋白质结合界面方面优于其各个分类器。
综合方法捕获了各个分类器的最佳性能,并提供了改进的接口预测。该方法稳健,即使个别分类器在特定查询蛋白上表现不佳,它也能很好地执行。这项工作表明,集成依赖于蛋白质不同结构特性的正交方法在界面预测方面的性能优于任何单个分类器。