Neuromuscular and Ataxias Research Group, Health Research Institute Hospital La Fe (IIS La Fe), Valencia, Spain.
Centre for Biomedical Network Research on Rare Diseases (CIBERER); U763, CB06/05/0091, Valencia, Spain.
Ann Neurol. 2022 Nov;92(5):793-806. doi: 10.1002/ana.26461. Epub 2022 Sep 7.
Duchenne muscular dystrophy (DMD) exon 45-55 deletion (del45-55) has been postulated as a model that could treat up to 60% of DMD patients, but the associated clinical variability and complications require clarification. We aimed to understand the phenotypes and potential modifying factors of this dystrophinopathy subset.
This cross-sectional, multicenter cohort study applied clinical and functional evaluation. Next generation sequencing was employed to identify intronic breakpoints and their impact on the Dp140 promotor, intronic long noncoding RNA, and regulatory splicing sequences. DMD modifiers (SPP1, LTBP4, ACTN3) and concomitant mutations were also assessed. Haplotypes were built using DMD single nucleotide polymorphisms. Dystrophin expression was evaluated via immunostaining, Western blotting, reverse transcription polymerase chain reaction (PCR), and droplet digital PCR in 9 muscle biopsies.
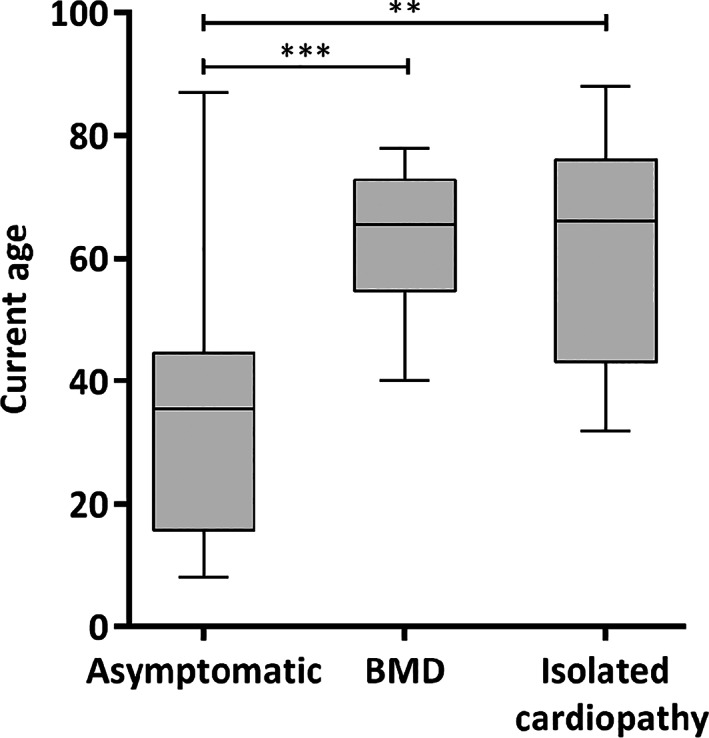
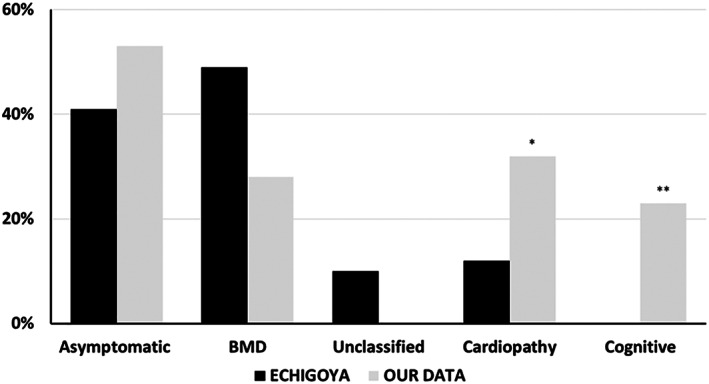
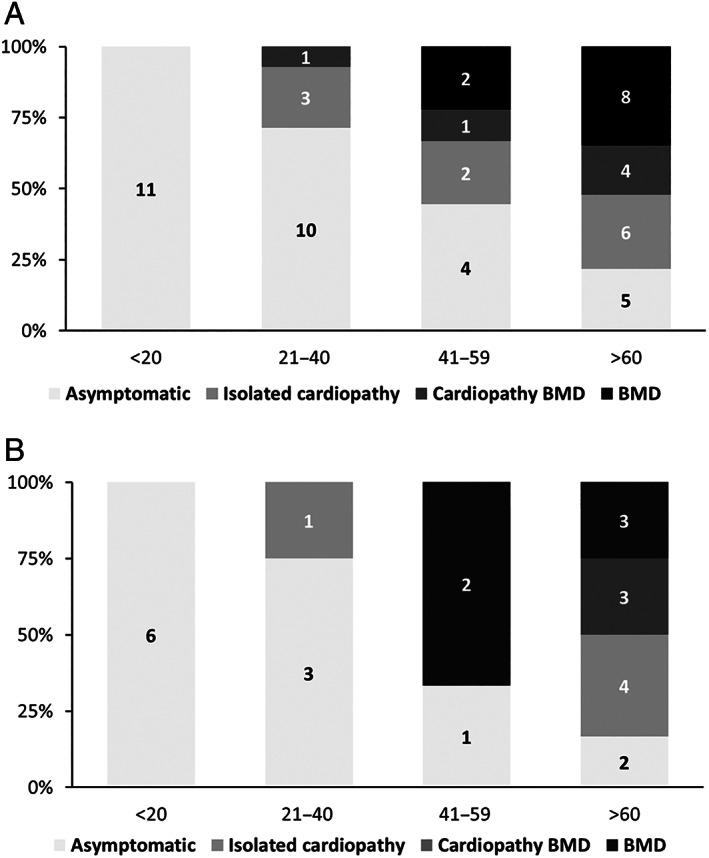
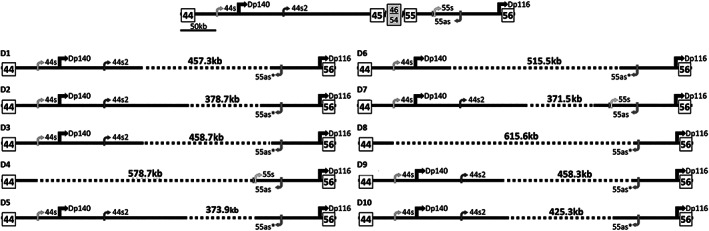
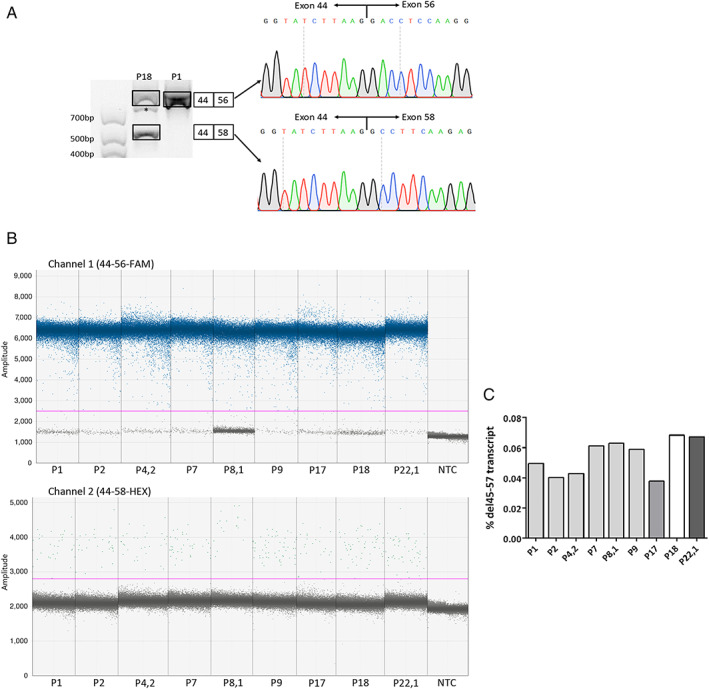
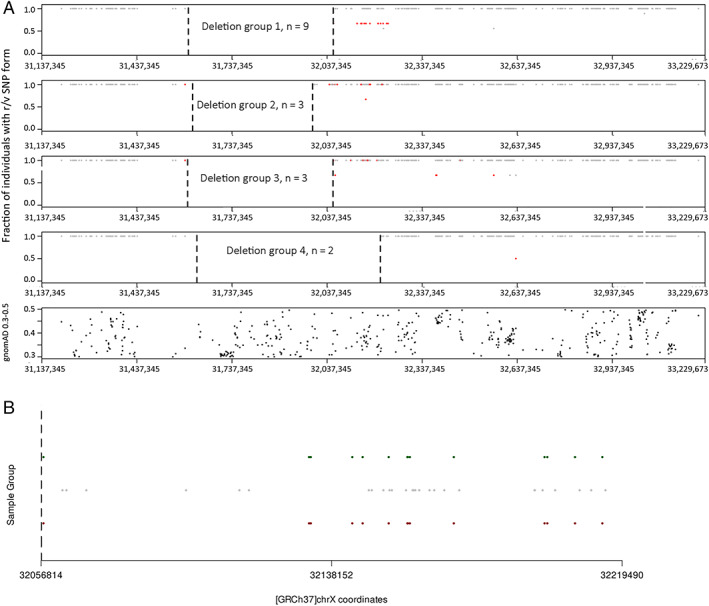
The series comprised 57 subjects (23 index) expressing Becker phenotype (28%), isolated cardiopathy (19%), and asymptomatic features (53%). Cognitive impairment occurred in 90% of children. Patients were classified according to 10 distinct index-case breakpoints; 4 of them were recurrent due to founder events. A specific breakpoint (D5) was associated with severity, but no significant effect was appreciated due to the changes in intronic sequences. All biopsies showed dystrophin expression of >67% and traces of alternative del45-57 transcript that were not deemed pathogenically relevant. Only the LTBP4 haplotype appeared associated the presence of cardiopathy among the explored extragenic factors.
We confirmed that del45-55 segregates a high proportion of benign phenotypes, severe cases, and isolated cardiac and cognitive presentations. Although some influence of the intronic breakpoint position and the LTBP4 modifier may exist, the pathomechanisms responsible for the phenotypic variability remain largely unresolved. ANN NEUROL 2022;92:793-806.
杜氏肌营养不良症(DMD)exon45-55 缺失(del45-55)被认为是一种可以治疗多达 60%的 DMD 患者的模型,但相关的临床变异性和并发症需要澄清。我们旨在了解这种抗肌萎缩蛋白病亚组的表型和潜在的修饰因子。
本项横断面、多中心队列研究采用临床和功能评估。下一代测序用于鉴定内含子断点及其对 Dp140 启动子、内含子长非编码 RNA 和调节性剪接序列的影响。还评估了 DMD 修饰因子(SPP1、LTBP4、ACTN3)和伴随突变。使用 DMD 单核苷酸多态性构建单体型。通过免疫染色、Western blot、反转录聚合酶链反应(PCR)和 9 例肌肉活检中的液滴数字 PCR 评估抗肌萎缩蛋白的表达。
该系列包括 57 名表达 Becker 表型(28%)、孤立性心脏病(19%)和无症状特征(53%)的受试者(23 名索引)。90%的儿童存在认知障碍。患者根据 10 个不同的索引病例断点进行分类;其中 4 个由于创始人事件而复发。一个特定的断点(D5)与严重程度相关,但由于内含子序列的变化,没有观察到显著的影响。所有活检均显示抗肌萎缩蛋白表达>67%,并存在微量的替代性 del45-57 转录物,但不被认为具有致病性。在探索的外显子因子中,只有 LTBP4 单体型似乎与心脏病的存在有关。
我们证实 del45-55 分离出很大比例的良性表型、严重病例以及孤立性心脏和认知表现。尽管内含子断点位置和 LTBP4 修饰因子的某些影响可能存在,但导致表型变异性的病理机制仍在很大程度上未得到解决。