Department of Molecular Cell Biology, Weizmann Institute of Science, 7610001 Rehovot, Israel.
Department of Computer Science and Applied Mathematics and Department of Molecular Cell Biology, Weizmann Institute of Science, 7610001 Rehovot, Israel.
Cell. 2022 Aug 18;185(17):3169-3185.e20. doi: 10.1016/j.cell.2022.06.049. Epub 2022 Jul 30.
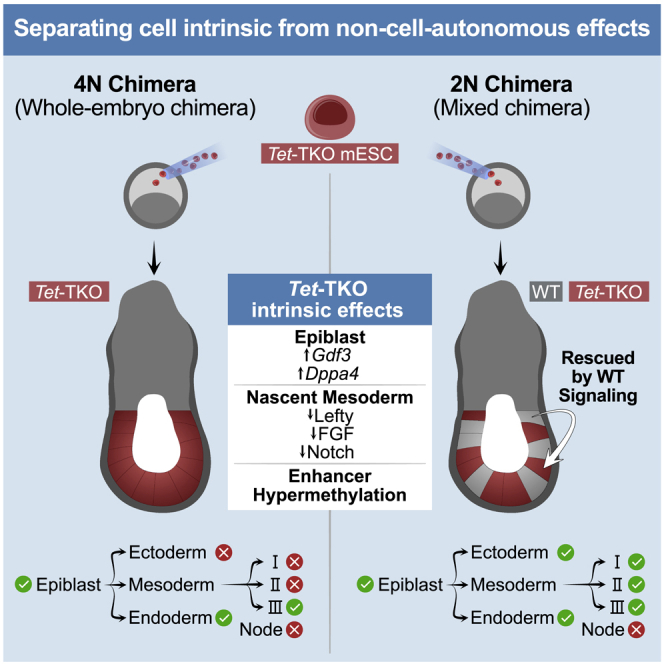
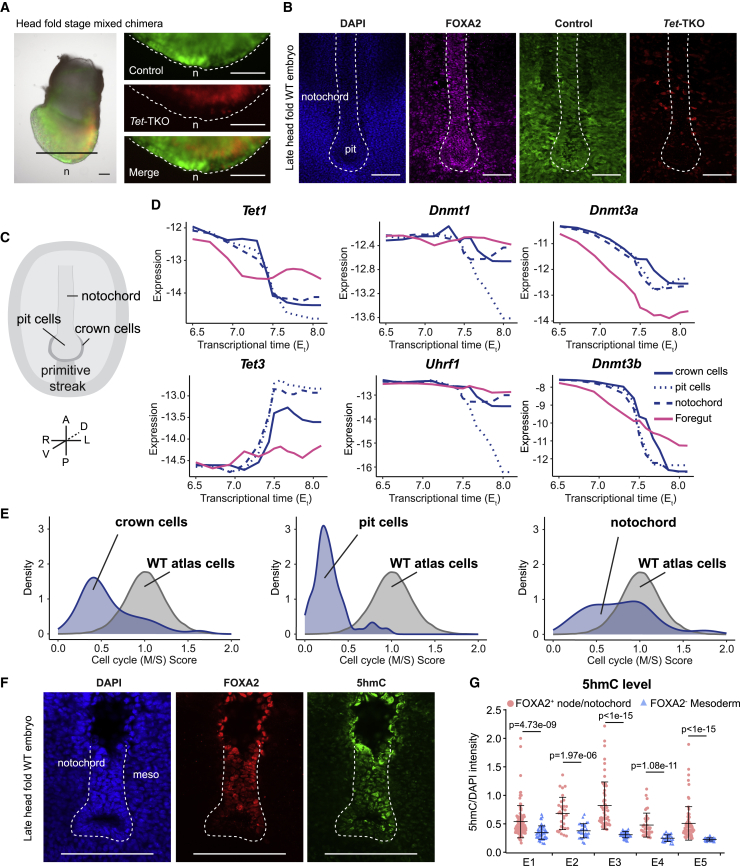
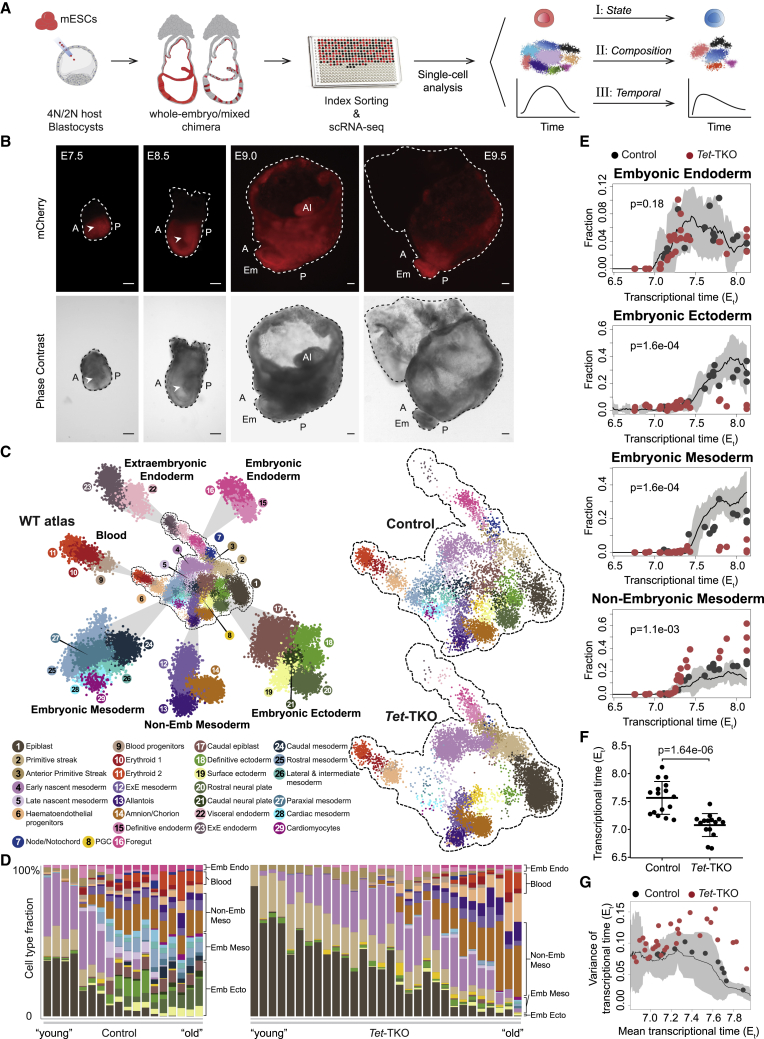
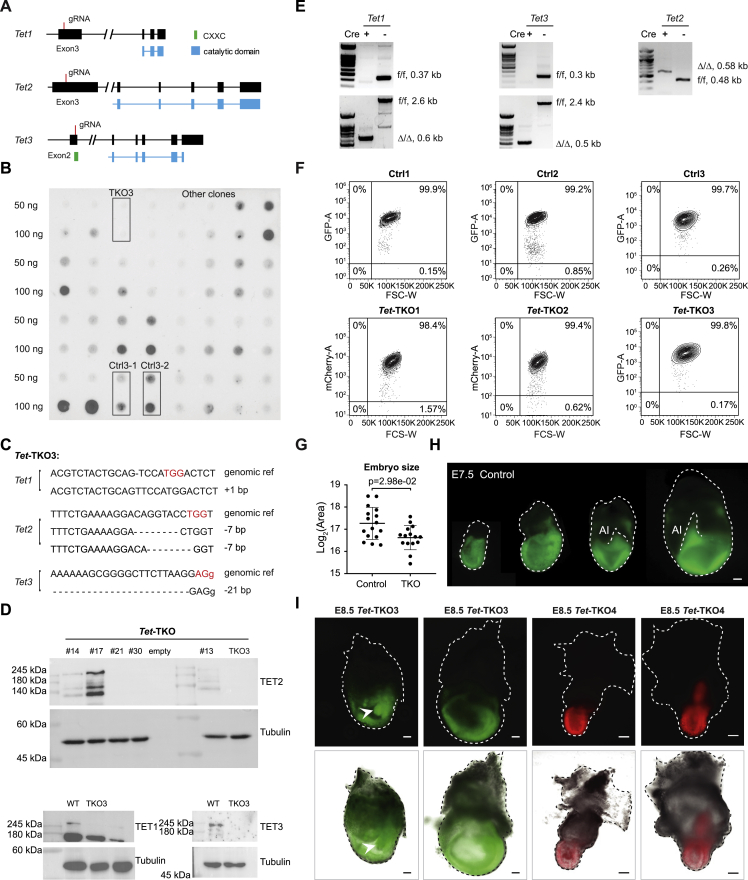
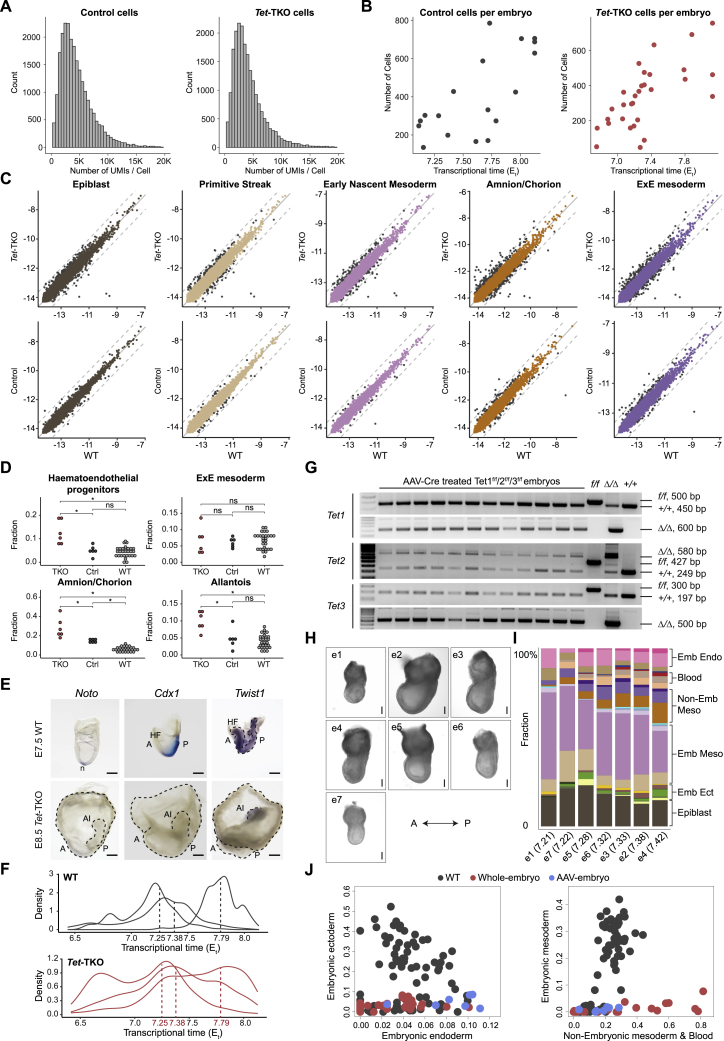
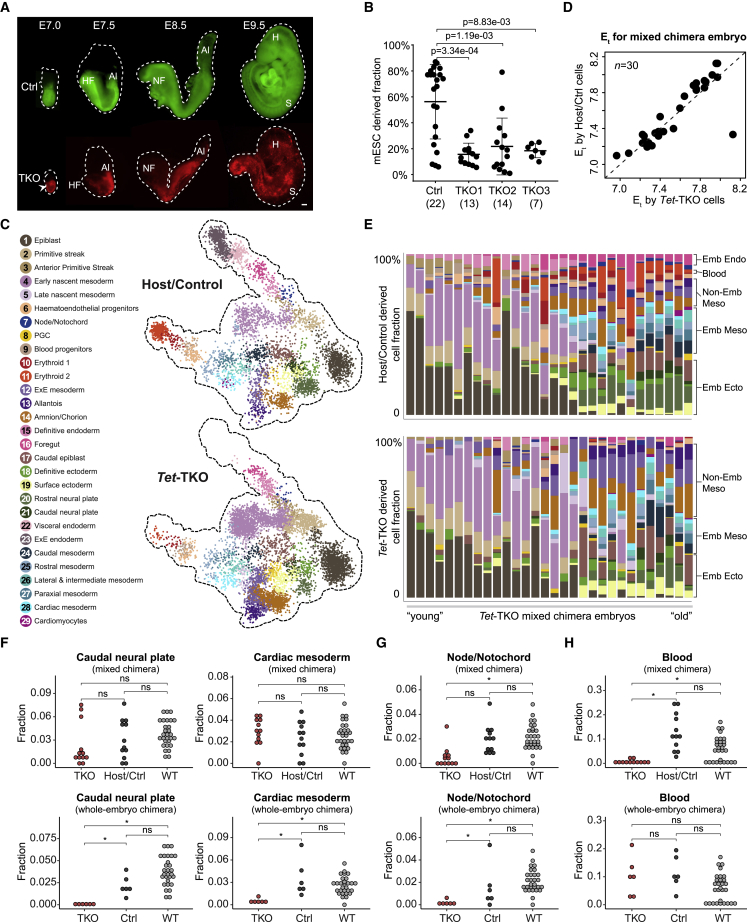
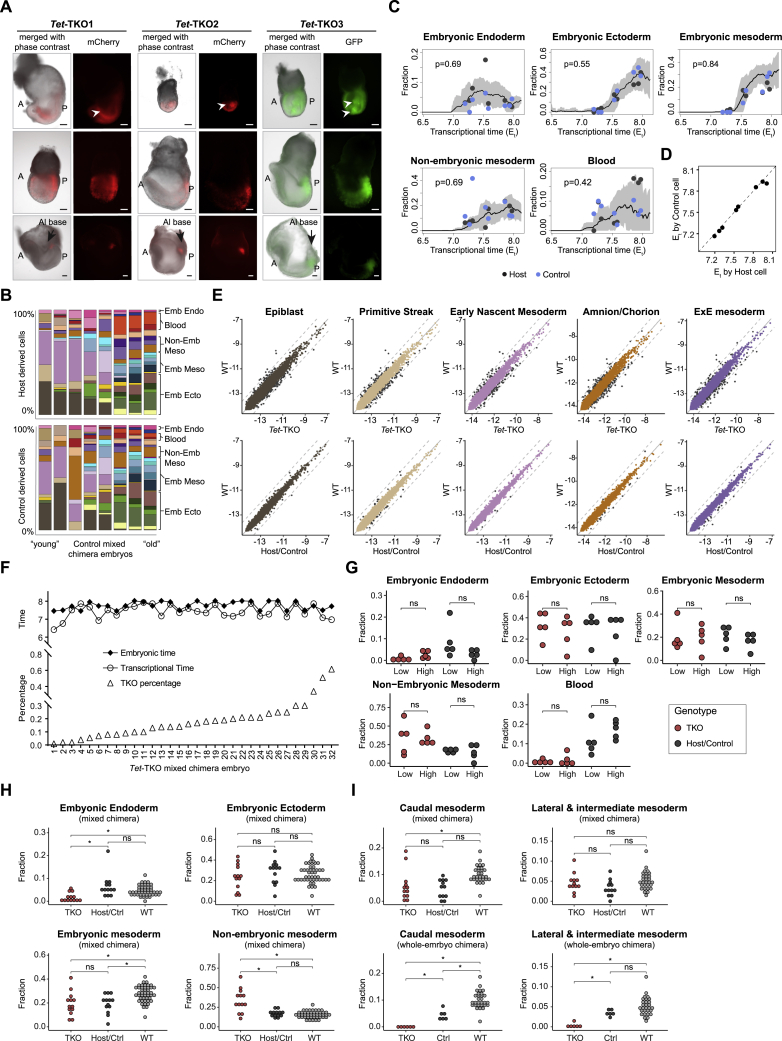
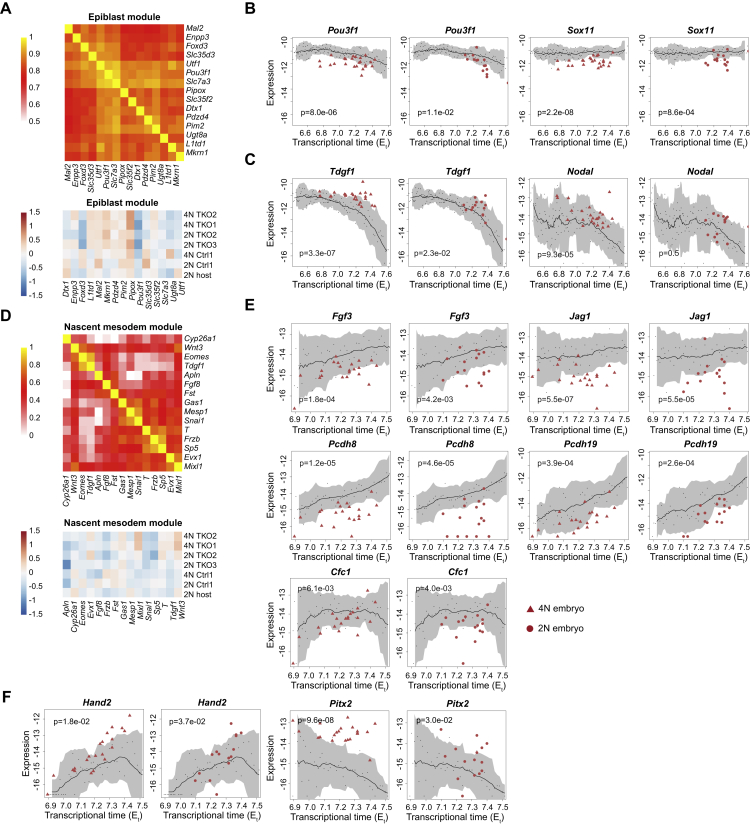
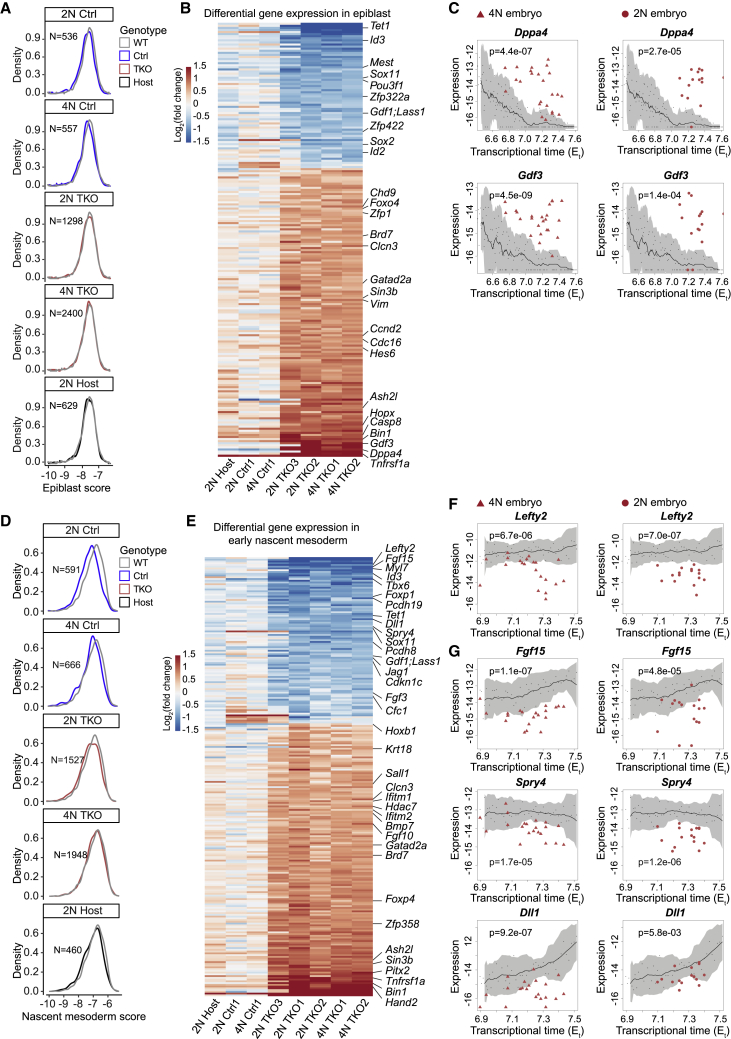
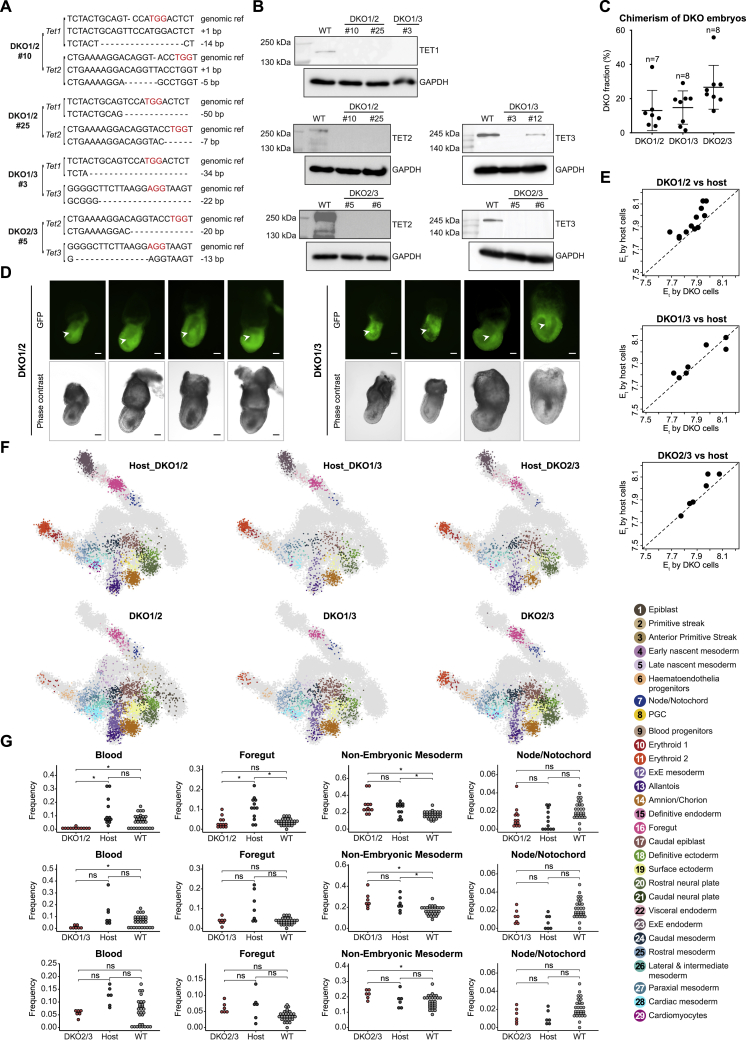
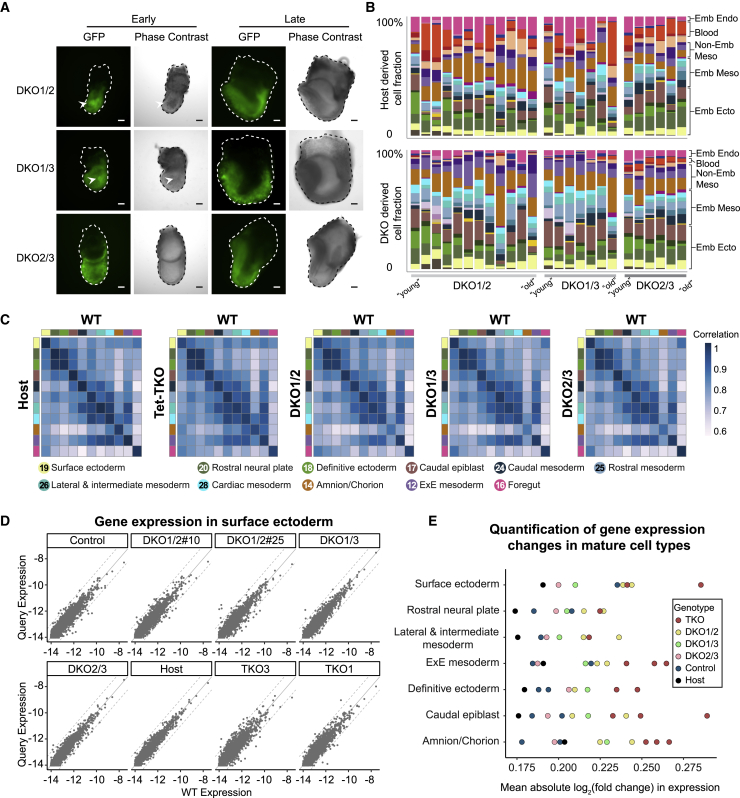
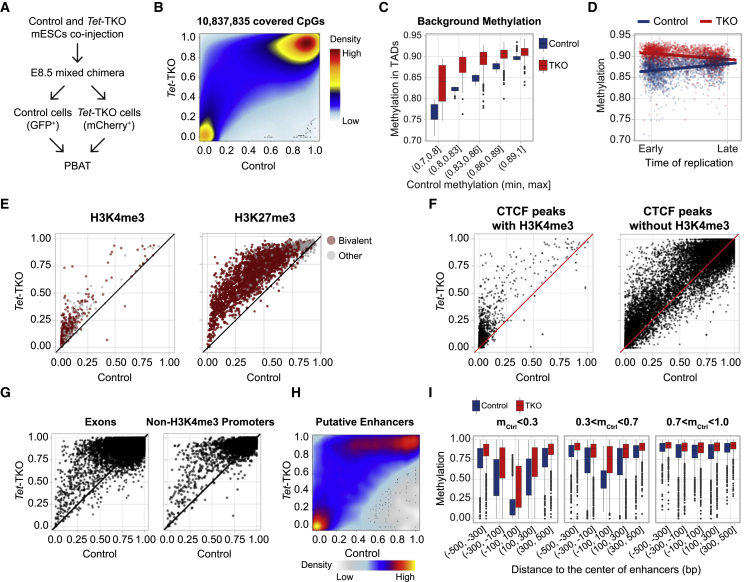
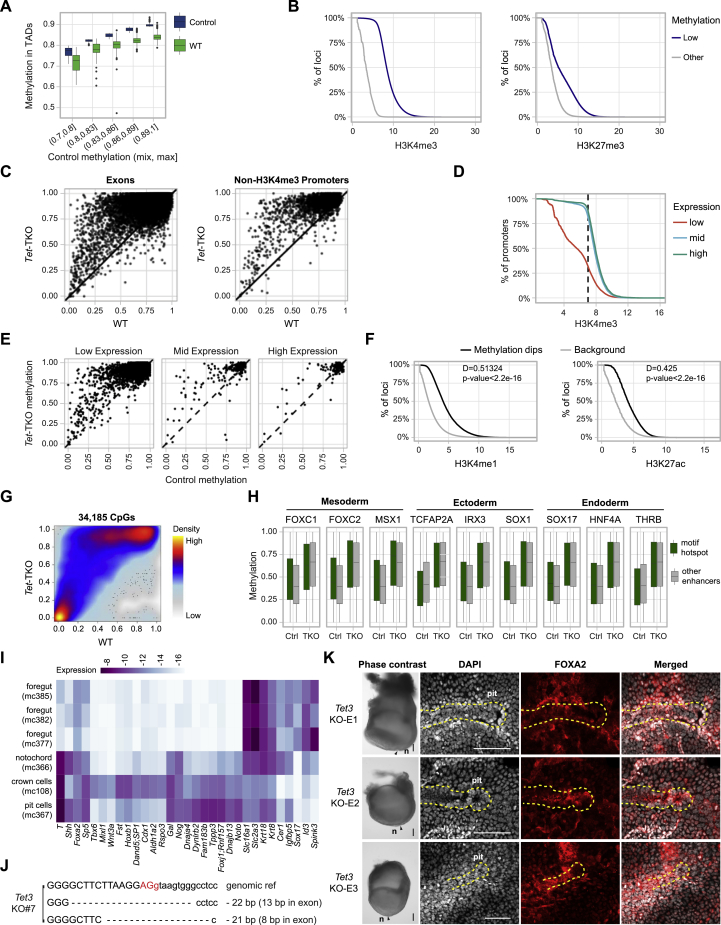
Mice deficient for all ten-eleven translocation (TET) genes exhibit early gastrulation lethality. However, separating cause and effect in such embryonic failure is challenging. To isolate cell-autonomous effects of TET loss, we used temporal single-cell atlases from embryos with partial or complete mutant contributions. Strikingly, when developing within a wild-type embryo, Tet-mutant cells retain near-complete differentiation potential, whereas embryos solely comprising mutant cells are defective in epiblast to ectoderm transition with degenerated mesoderm potential. We map de-repressions of early epiblast factors (e.g., Dppa4 and Gdf3) and failure to activate multiple signaling from nascent mesoderm (Lefty, FGF, and Notch) as likely cell-intrinsic drivers of TET loss phenotypes. We further suggest loss of enhancer demethylation as the underlying mechanism. Collectively, our work demonstrates an unbiased approach for defining intrinsic and extrinsic embryonic gene function based on temporal differentiation atlases and disentangles the intracellular effects of the demethylation machinery from its broader tissue-level ramifications.
所有十号染色体缺失的双微体 2(TET)基因缺失的小鼠表现出早期原肠胚致死。然而,在这种胚胎衰竭中区分因果关系具有挑战性。为了分离 TET 缺失的细胞自主性影响,我们使用了具有部分或完全突变贡献的胚胎的时间单细胞图谱。引人注目的是,当在野生型胚胎中发育时,Tet 突变细胞保留几乎完整的分化潜能,而仅由突变细胞组成的胚胎在胚外胚层到外胚层的过渡中存在缺陷,中胚层潜能退化。我们将早期内胚层因子(例如 Dppa4 和 Gdf3)的去抑制和新生中胚层中多个信号(Lefty、FGF 和 Notch)的激活失败映射为 TET 缺失表型的可能细胞内在驱动因素。我们进一步提出增强子去甲基化的丧失作为潜在机制。总之,我们的工作展示了一种基于时间分化图谱定义内在和外在胚胎基因功能的无偏方法,并将去甲基化机制的细胞内效应与其更广泛的组织水平影响区分开来。