Department of Computer Science, University of Manchester, Manchester, UK.
Department of Computer Science, Taif University, Taif, Saudi Arabia.
BMC Bioinformatics. 2022 Aug 6;23(1):323. doi: 10.1186/s12859-022-04787-8.
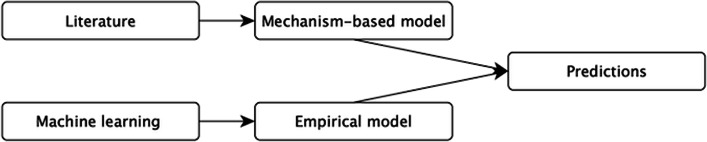
A key problem in bioinformatics is that of predicting gene expression levels. There are two broad approaches: use of mechanistic models that aim to directly simulate the underlying biology, and use of machine learning (ML) to empirically predict expression levels from descriptors of the experiments. There are advantages and disadvantages to both approaches: mechanistic models more directly reflect the underlying biological causation, but do not directly utilize the available empirical data; while ML methods do not fully utilize existing biological knowledge.
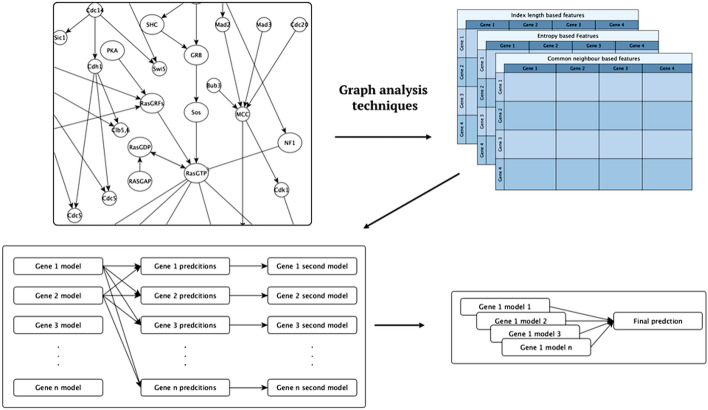
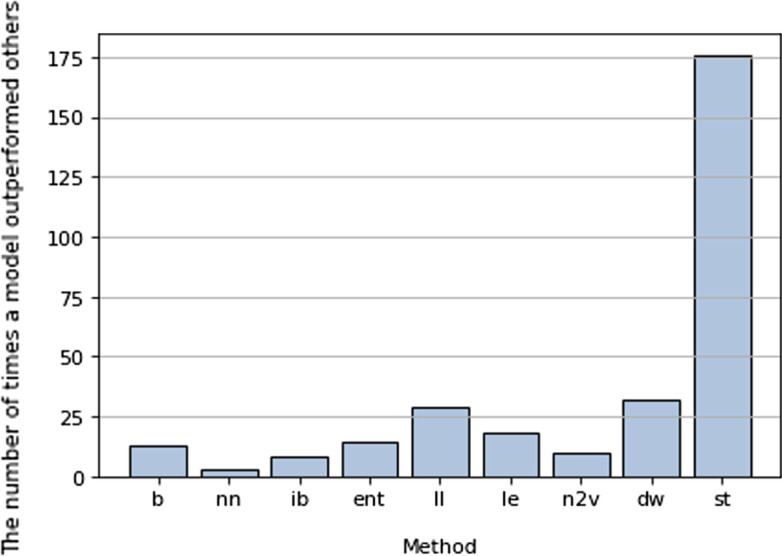
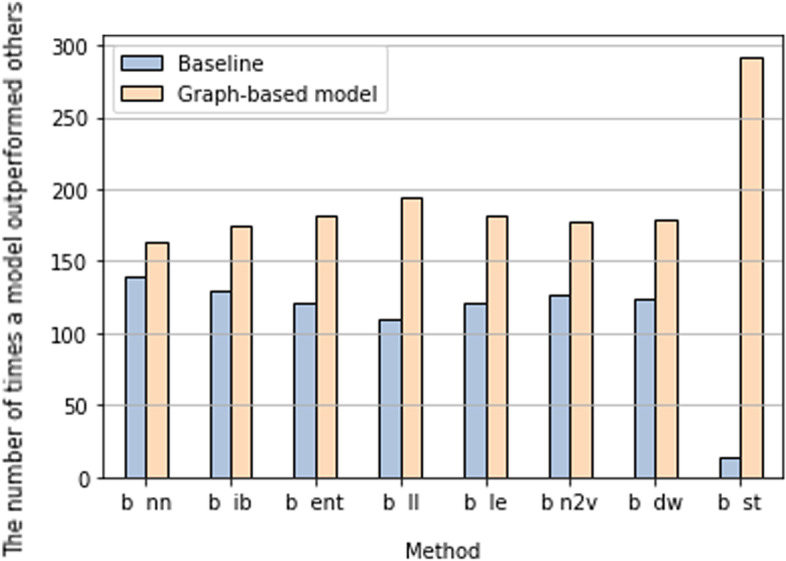
Here, we investigate overcoming these disadvantages by integrating mechanistic cell signalling models with ML. Our approach to integration is to augment ML with similarity features (attributes) computed from cell signalling models. Seven sets of different similarity feature were generated using graph theory. Each set of features was in turn used to learn multi-target regression models. All the features have significantly improved accuracy over the baseline model - without the similarity features. Finally, the seven multi-target regression models were stacked together to form an overall prediction model that was significantly better than the baseline on 95% of genes on an independent test set. The similarity features enable this stacking model to provide interpretable knowledge about cancer, e.g. the role of ERBB3 in the MCF7 breast cancer cell line.
Integrating mechanistic models as graphs helps to both improve the predictive results of machine learning models, and to provide biological knowledge about genes that can help in building state-of-the-art mechanistic models.
生物信息学中的一个关键问题是预测基因表达水平。有两种广泛的方法:使用旨在直接模拟基础生物学的机械模型,以及使用机器学习 (ML) 从实验描述符中经验预测表达水平。这两种方法都有优点和缺点:机械模型更直接地反映了潜在的生物学因果关系,但不能直接利用可用的经验数据;而 ML 方法没有充分利用现有的生物学知识。
在这里,我们通过将机械细胞信号模型与 ML 集成来研究克服这些缺点。我们的集成方法是用从细胞信号模型计算出的相似性特征(属性)来增强 ML。使用图论生成了七组不同的相似性特征。每组特征依次用于学习多目标回归模型。所有特征的准确性都明显优于没有相似性特征的基线模型。最后,将这七个多目标回归模型堆叠在一起,形成一个整体预测模型,在独立测试集上 95%的基因上的表现明显优于基线模型。相似性特征使这种堆叠模型能够提供有关癌症的可解释知识,例如 ERBB3 在 MCF7 乳腺癌细胞系中的作用。
将机械模型集成为图有助于提高机器学习模型的预测结果,并提供有关基因的生物学知识,这有助于构建最先进的机械模型。