Kwapien Karolina, Nittinger Eva, He Jiazhen, Margreitter Christian, Voronov Alexey, Tyrchan Christian
Medicinal Chemistry, Research and Early Development, Respiratory and Immunology (R&I), BioPharmaceuticals R&D, AstraZeneca, Gothenburg 431 83, Sweden.
Molecular AI, Discovery Sciences, R&D, AstraZeneca, Gothenburg 431 83, Sweden.
ACS Omega. 2022 Jul 19;7(30):26573-26581. doi: 10.1021/acsomega.2c02738. eCollection 2022 Aug 2.
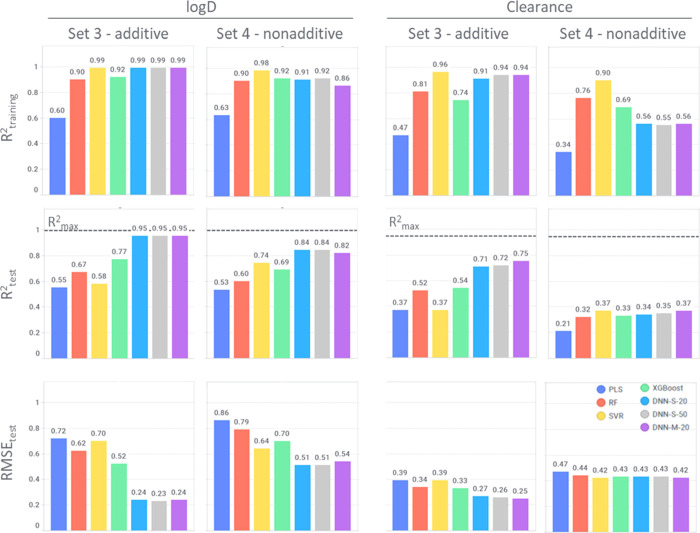
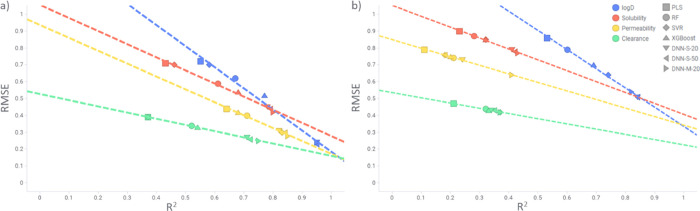
Matched molecular pairs (MMPs) are nowadays a commonly applied concept in drug design. They are used in many computational tools for structure-activity relationship analysis, biological activity prediction, or optimization of physicochemical properties. However, until now it has not been shown in a rigorous way that MMPs, that is, changing only one substituent between two molecules, can be predicted with higher accuracy and precision in contrast to any other chemical compound pair. It is expected that any model should be able to predict such a defined change with high accuracy and reasonable precision. In this study, we examine the predictability of four classical properties relevant for drug design ranging from simple physicochemical parameters (log and solubility) to more complex cell-based ones (permeability and clearance), using different data sets and machine learning algorithms. Our study confirms that additive data are the easiest to predict, which highlights the importance of recognition of nonadditivity events and the challenging complexity of predicting properties in case of scaffold hopping. Despite deep learning being well suited to model nonlinear events, these methods do not seem to be an exception of this observation. Though they are in general performing better than classical machine learning methods, this leaves the field with a still standing challenge.
匹配分子对(MMPs)如今是药物设计中常用的概念。它们被用于许多计算工具中进行构效关系分析、生物活性预测或物理化学性质优化。然而,到目前为止,尚未有严格的研究表明,与任何其他化合物对相比,仅改变两个分子间的一个取代基的MMPs能够以更高的准确性和精确性被预测。预计任何模型都应能够以高准确性和合理的精确性预测这种特定的变化。在本研究中,我们使用不同的数据集和机器学习算法,研究了与药物设计相关的四种经典性质的可预测性,这些性质涵盖了从简单的物理化学参数(logP和溶解度)到更复杂的基于细胞的参数(渗透性和清除率)。我们的研究证实,加和性数据最容易预测,这凸显了识别非加和性事件的重要性以及在骨架跃迁情况下预测性质的挑战性复杂性。尽管深度学习非常适合对非线性事件进行建模,但这些方法似乎也不例外。虽然它们总体上比经典机器学习方法表现更好,但这仍然给该领域留下了一个悬而未决的挑战。