Institute for Complex Molecular Systems and Dept. Biomedical Engineering, Eindhoven University of Technology, 5612AZEindhoven, The Netherlands.
Centre for Living Technologies, Alliance TU/e, WUR, UU, UMC Utrecht, 3584CBUtrecht, The Netherlands.
J Chem Inf Model. 2022 Dec 12;62(23):5938-5951. doi: 10.1021/acs.jcim.2c01073. Epub 2022 Dec 1.
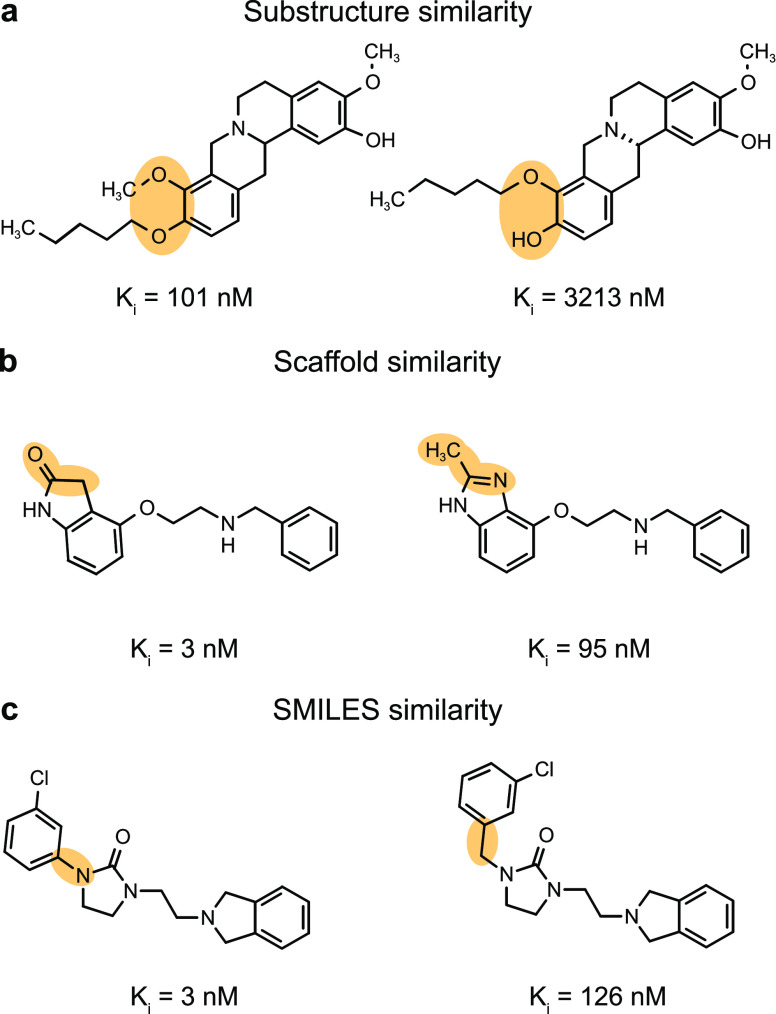
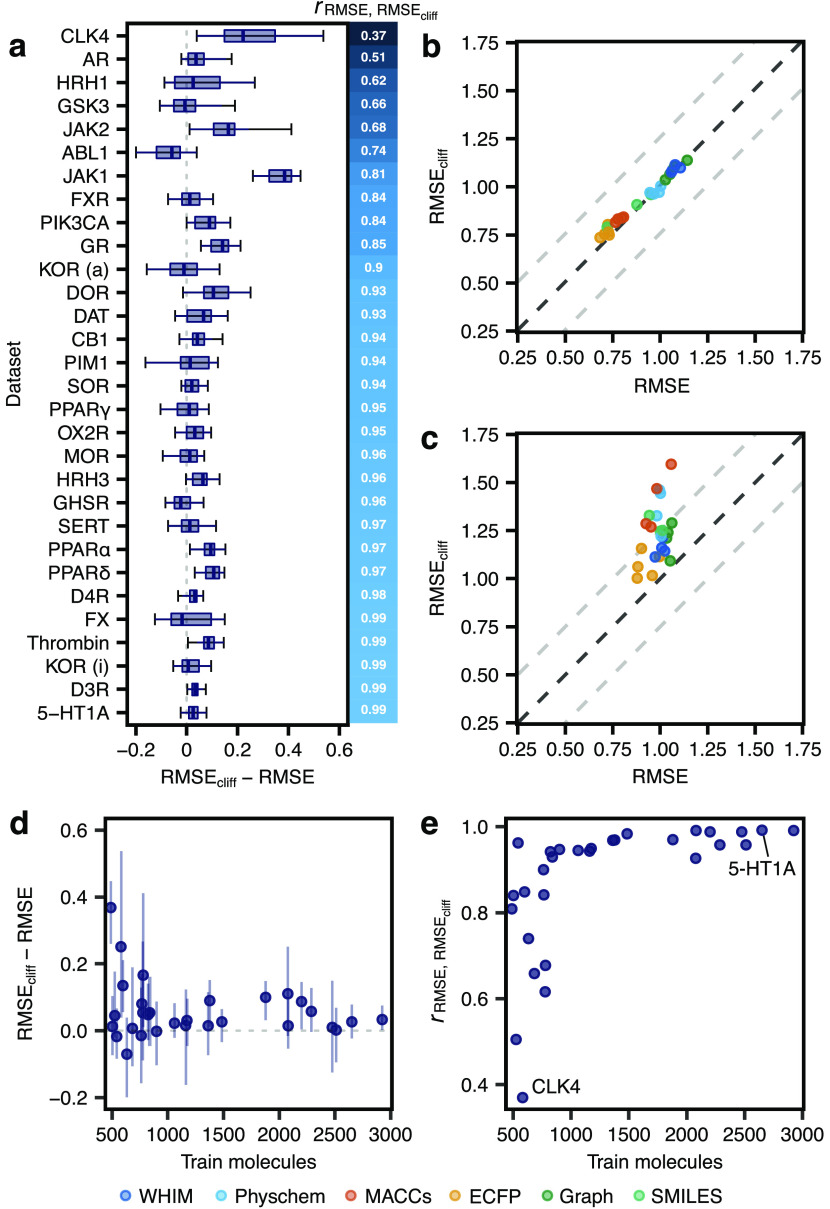
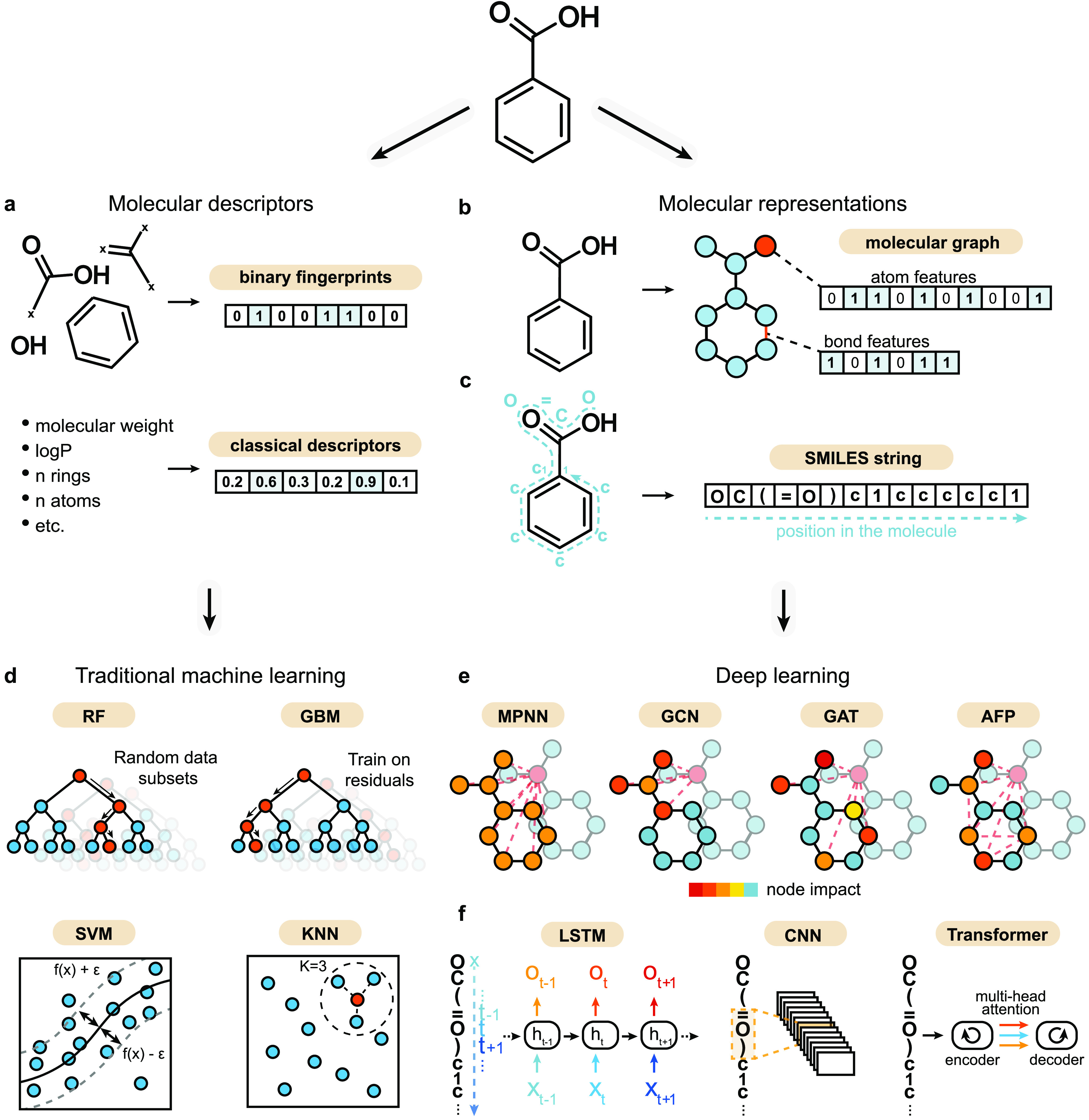
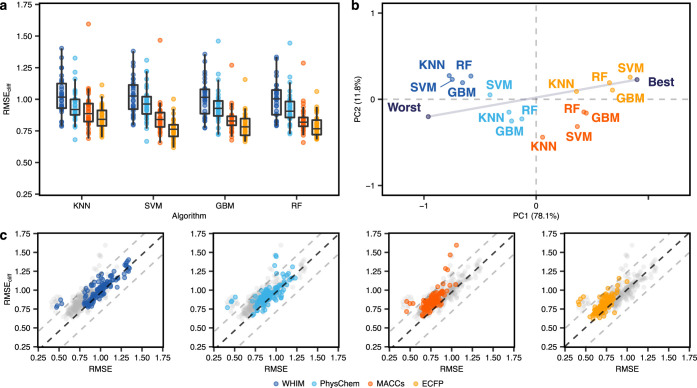
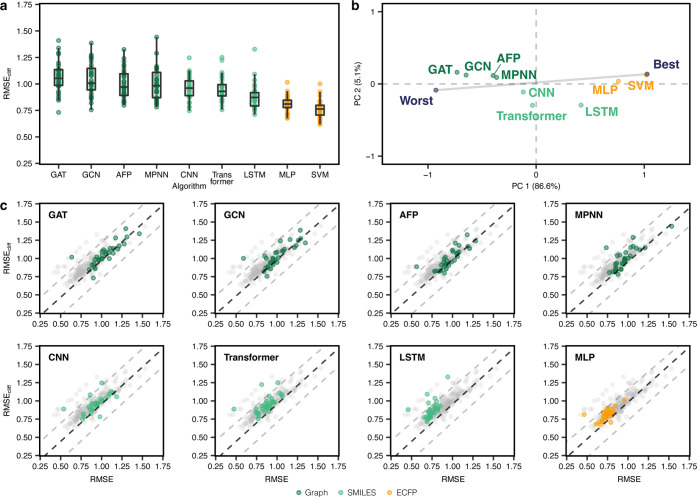
Machine learning has become a crucial tool in drug discovery and chemistry at large, , to predict molecular properties, such as bioactivity, with high accuracy. However, activity cliffs─pairs of molecules that are highly similar in their structure but exhibit large differences in potency─have received limited attention for their effect on model performance. Not only are these edge cases informative for molecule discovery and optimization but also models that are well equipped to accurately predict the potency of activity cliffs have increased potential for prospective applications. Our work aims to fill the current knowledge gap on best-practice machine learning methods in the presence of activity cliffs. We benchmarked a total of 24 machine and deep learning approaches on curated bioactivity data from 30 macromolecular targets for their performance on activity cliff compounds. While all methods struggled in the presence of activity cliffs, machine learning approaches based on molecular descriptors outperformed more complex deep learning methods. Our findings highlight large case-by-case differences in performance, advocating for (a) the inclusion of dedicated "activity-cliff-centered" metrics during model development and evaluation and (b) the development of novel algorithms to better predict the properties of activity cliffs. To this end, the methods, metrics, and results of this study have been encapsulated into an open-access benchmarking platform named MoleculeACE (Activity Cliff Estimation, available on GitHub at: https://github.com/molML/MoleculeACE). MoleculeACE is designed to steer the community toward addressing the pressing but overlooked limitation of molecular machine learning models posed by activity cliffs.
机器学习已经成为药物发现和整个化学领域的重要工具,可用于高精度地预测分子性质,如生物活性。然而,活性悬崖(结构高度相似但活性差异很大的分子对)对模型性能的影响受到的关注有限。这些边缘情况不仅对分子发现和优化具有启示意义,而且能够准确预测活性悬崖活性的模型也更有可能应用于前瞻性应用。我们的工作旨在填补当前关于存在活性悬崖时机器学习方法的最佳实践的知识空白。我们总共在 30 个大分子靶标上的生物活性数据上对 24 种机器和深度学习方法进行了基准测试,以评估它们对活性悬崖化合物的性能。虽然所有方法在活性悬崖存在的情况下都存在困难,但基于分子描述符的机器学习方法的性能优于更复杂的深度学习方法。我们的研究结果突出了性能的大案例差异,主张在模型开发和评估过程中纳入专门的“活性悬崖中心”指标,以及开发更好地预测活性悬崖性质的新算法。为此,这项研究的方法、指标和结果已经被封装到一个名为 MoleculeACE(Activity Cliff Estimation,可在 GitHub 上获得:https://github.com/molML/MoleculeACE)的开放访问基准测试平台中。MoleculeACE 的设计旨在引导社区解决由活性悬崖引起的分子机器学习模型的紧迫但被忽视的限制。