Institute for the Advanced Study of Human Biology, Kyoto University Institute for Advanced Study, Kyoto University, Kyoto, Japan.
Department of Anatomy and Cell Biology, Graduate School of Medicine, Kyoto University, Kyoto, Japan.
Life Sci Alliance. 2022 Aug 9;5(12):e202201591. doi: 10.26508/lsa.202201591.
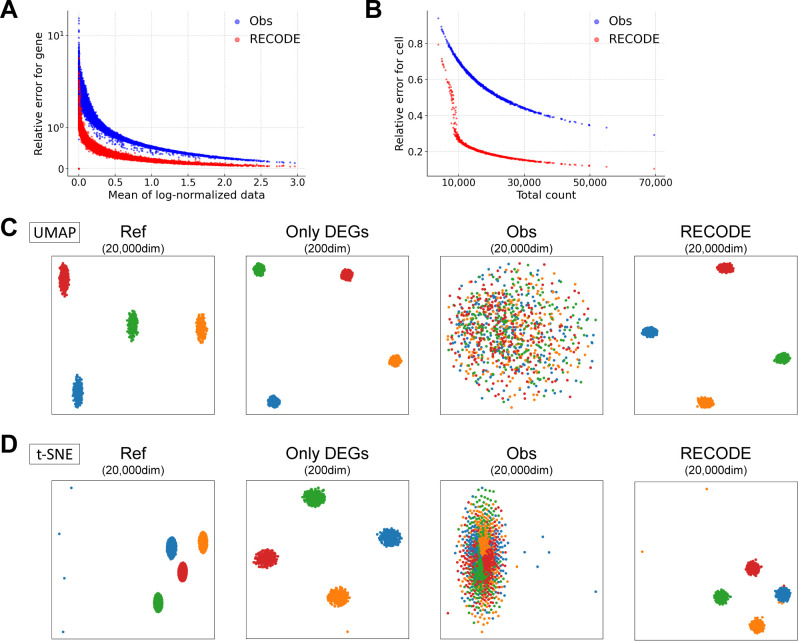
Single-cell RNA sequencing (scRNA-seq) can determine gene expression in numerous individual cells simultaneously, promoting progress in the biomedical sciences. However, scRNA-seq data are high-dimensional with substantial technical noise, including dropouts. During analysis of scRNA-seq data, such noise engenders a statistical problem known as the curse of dimensionality (COD). Based on high-dimensional statistics, we herein formulate a noise reduction method, RECODE (resolution of the curse of dimensionality), for high-dimensional data with random sampling noise. We show that RECODE consistently resolves COD in relevant scRNA-seq data with unique molecular identifiers. RECODE does not involve dimension reduction and recovers expression values for all genes, including lowly expressed genes, realizing precise delineation of cell fate transitions and identification of rare cells with all gene information. Compared with representative imputation methods, RECODE employs different principles and exhibits superior overall performance in cell-clustering, expression value recovery, and single-cell-level analysis. The RECODE algorithm is parameter-free, data-driven, deterministic, and high-speed, and its applicability can be predicted based on the variance normalization performance. We propose RECODE as a powerful strategy for preprocessing noisy high-dimensional data.
单细胞 RNA 测序(scRNA-seq)可以同时确定大量单个细胞中的基因表达情况,从而推动生物医学科学的发展。然而,scRNA-seq 数据具有高度的维度,并且存在大量的技术噪声,包括缺失值。在分析 scRNA-seq 数据时,这种噪声会产生一个称为维度灾难(COD)的统计问题。基于高维统计学,我们在此提出了一种针对随机采样噪声的高维数据降噪方法,RECODE(维度灾难的解决)。我们表明,RECODE 可以通过独特的分子标识符一致地解决相关 scRNA-seq 数据中的 COD。RECODE 不涉及降维,并且可以恢复所有基因的表达值,包括低表达基因,从而实现细胞命运转变的精确描绘,并利用所有基因信息识别稀有细胞。与代表性的插补方法相比,RECODE 采用了不同的原理,在细胞聚类、表达值恢复和单细胞水平分析方面具有卓越的整体性能。RECODE 算法是无参数、数据驱动、确定性和高速的,其适用性可以根据方差归一化性能进行预测。我们提出 RECODE 作为一种强大的预处理噪声高维数据的策略。