Computational Biology Program, Ontario Institute for Cancer Research, Toronto, Canada.
Department of Medical Biophysics, University of Toronto, Toronto, Canada.
PLoS Comput Biol. 2022 Aug 10;18(8):e1010393. doi: 10.1371/journal.pcbi.1010393. eCollection 2022 Aug.
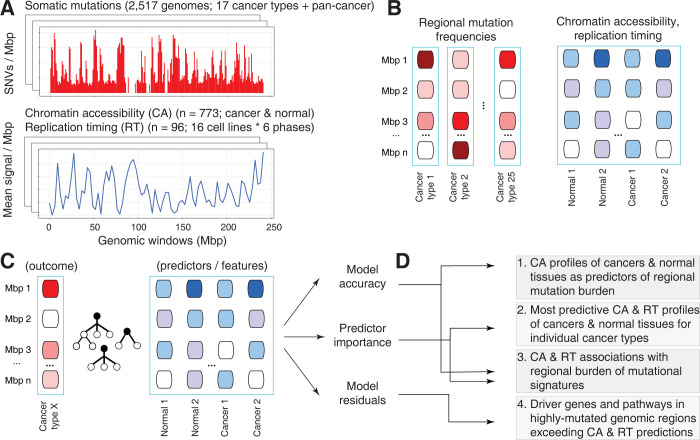
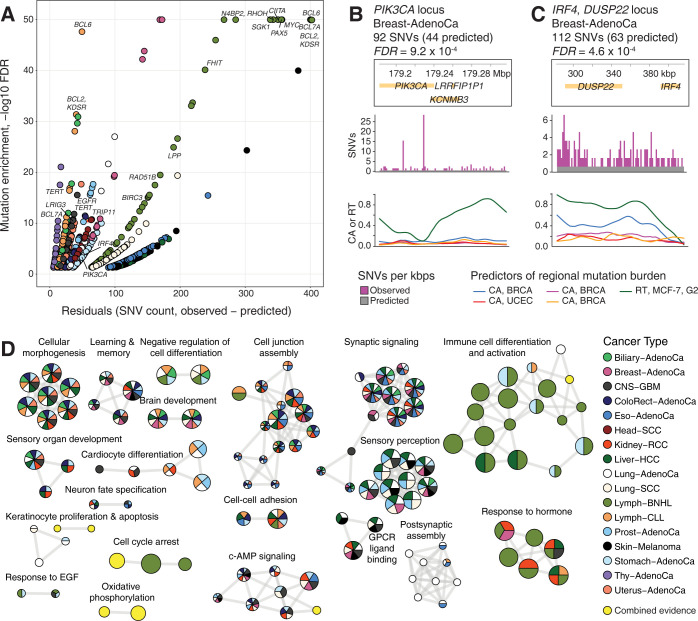
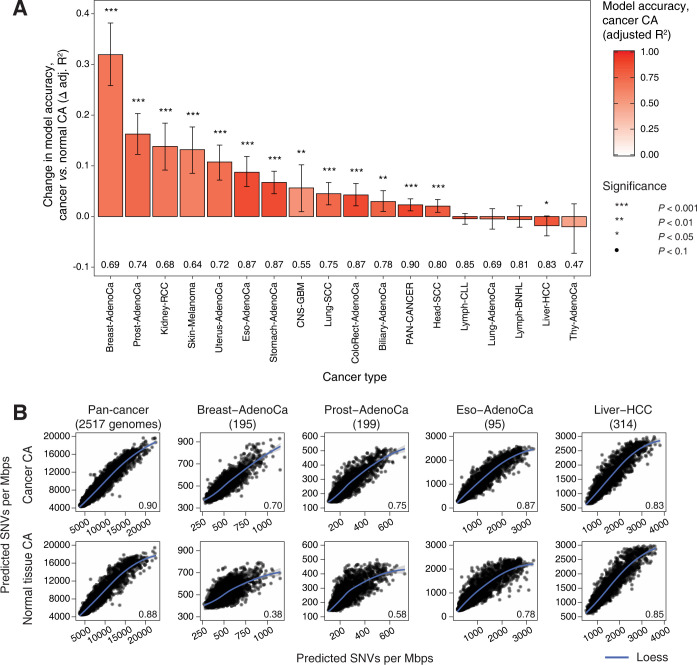
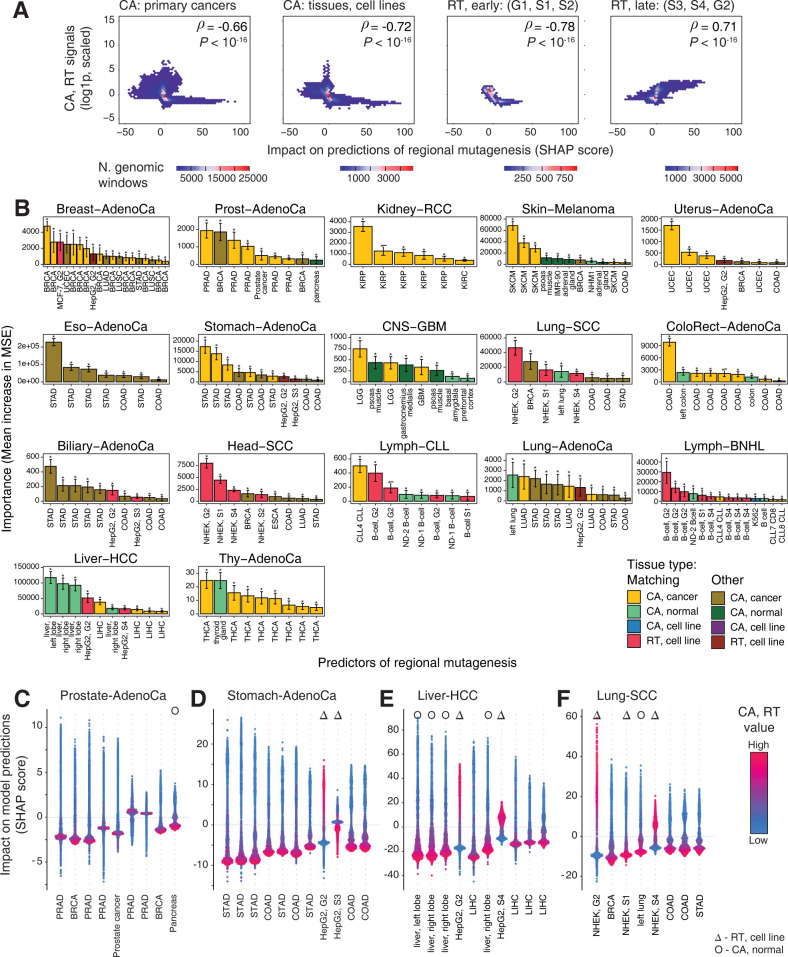
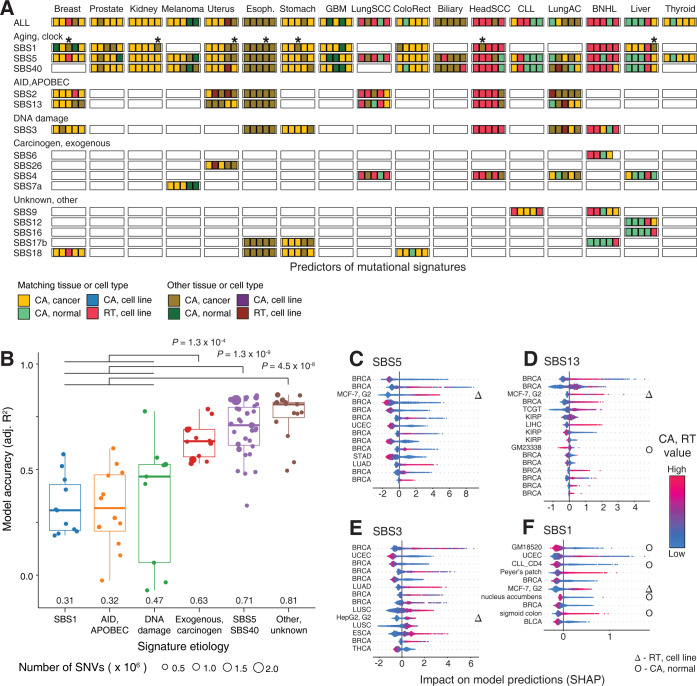
Somatic mutations in cancer genomes are associated with DNA replication timing (RT) and chromatin accessibility (CA), however these observations are based on normal tissues and cell lines while primary cancer epigenomes remain uncharacterised. Here we use machine learning to model megabase-scale mutation burden in 2,500 whole cancer genomes and 17 cancer types via a compendium of 900 CA and RT profiles covering primary cancers, normal tissues, and cell lines. CA profiles of primary cancers, rather than those of normal tissues, are most predictive of regional mutagenesis in most cancer types. Feature prioritisation shows that the epigenomes of matching cancer types and organ systems are often the strongest predictors of regional mutation burden, highlighting disease-specific associations of mutational processes. The genomic distributions of mutational signatures are also shaped by the epigenomes of matched cancer and tissue types, with SBS5/40, carcinogenic and unknown signatures most accurately predicted by our models. In contrast, fewer associations of RT and regional mutagenesis are found. Lastly, the models highlight genomic regions with overrepresented mutations that dramatically exceed epigenome-derived expectations and show a pan-cancer convergence to genes and pathways involved in development and oncogenesis, indicating the potential of this approach for coding and non-coding driver discovery. The association of regional mutational processes with the epigenomes of primary cancers suggests that the landscape of passenger mutations is predominantly shaped by the epigenomes of cancer cells after oncogenic transformation.
癌症基因组中的体细胞突变与 DNA 复制时间 (RT) 和染色质可及性 (CA) 有关,但这些观察结果基于正常组织和细胞系,而原发性癌症表观基因组尚未得到描述。在这里,我们通过涵盖原发性癌症、正常组织和细胞系的 900 个 CA 和 RT 图谱的摘要,使用机器学习对 2500 个全癌症基因组和 17 种癌症类型进行了兆碱基规模的突变负担建模。在大多数癌症类型中,原发性癌症的 CA 图谱而不是正常组织的 CA 图谱最能预测区域诱变。特征优先级显示,匹配癌症类型和器官系统的表观基因组通常是区域突变负担的最强预测因子,突出了突变过程的疾病特异性关联。突变特征的基因组分布也受到匹配癌症和组织类型的表观基因组的影响,我们的模型最能准确预测 SBS5/40、致癌和未知特征。相比之下,RT 和区域诱变的关联较少。最后,该模型突出了具有代表性突变的基因组区域,这些突变明显超过了基于表观基因组的预期,并且在癌症和发育及致癌途径中涉及的基因和途径上表现出泛癌症收敛,表明该方法在编码和非编码驱动基因发现方面具有潜力。区域突变过程与原发性癌症表观基因组的关联表明,乘客突变的景观主要是由致癌转化后癌细胞的表观基因组塑造的。