Kang Hyeunseok, Goo Sungwoo, Lee Hyunjung, Chae Jung-Woo, Yun Hwi-Yeol, Jung Sangkeun
Department of Bio-AI Convergence, Chungnam National University, Daejeon 34134, Korea.
College of Pharmacy, Chungnam National University, Daejeon 34134, Korea.
Pharmaceutics. 2022 Aug 16;14(8):1710. doi: 10.3390/pharmaceutics14081710.
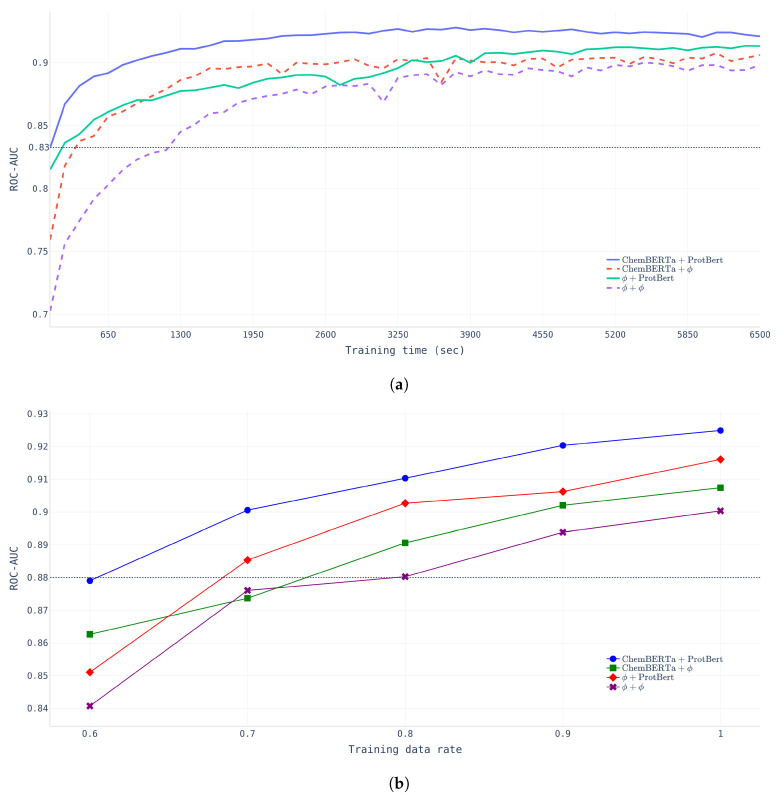
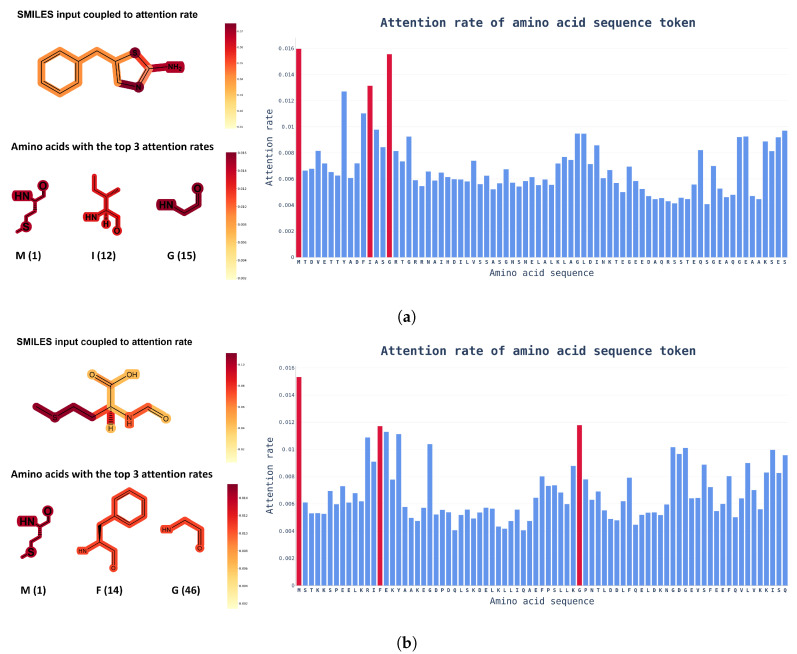
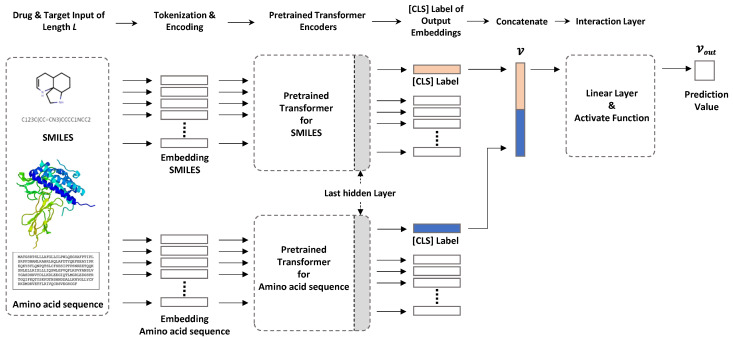
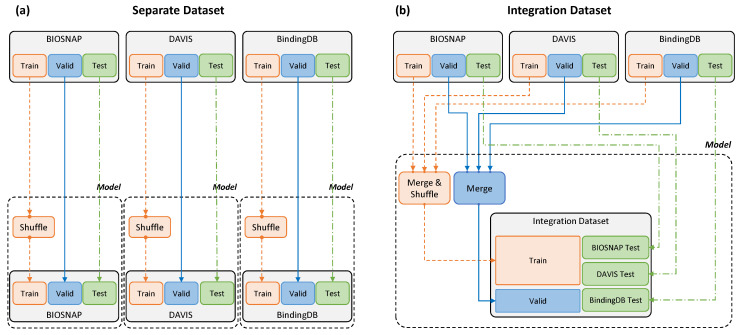
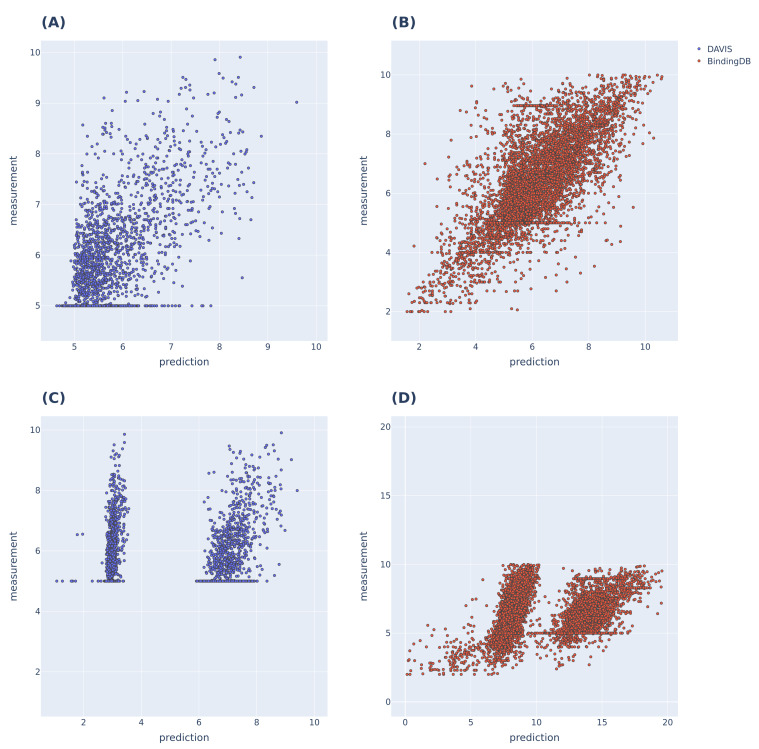
The identification of optimal drug candidates is very important in drug discovery. Researchers in biology and computational sciences have sought to use machine learning (ML) to efficiently predict drug-target interactions (DTIs). In recent years, according to the emerging usefulness of pretrained models in natural language process (NLPs), pretrained models are being developed for chemical compounds and target proteins. This study sought to improve DTI predictive models using a Bidirectional Encoder Representations from the Transformers (BERT)-pretrained model, ChemBERTa, for chemical compounds. Pretraining features the use of a simplified molecular-input line-entry system (SMILES). We also employ the pretrained ProBERT for target proteins (pretraining employed the amino acid sequences). The BIOSNAP, DAVIS, and BindingDB databases (DBs) were used (alone or together) for learning. The final model, taught by both ChemBERTa and ProtBert and the integrated DBs, afforded the best DTI predictive performance to date based on the receiver operating characteristic area under the curve (AUC) and precision-recall-AUC values compared with previous models. The performance of the final model was verified using a specific case study on 13 pairs of subtrates and the metabolic enzyme cytochrome P450 (CYP). The final model afforded excellent DTI prediction. As the real-world interactions between drugs and target proteins are expected to exhibit specific patterns, pretraining with ChemBERTa and ProtBert could teach such patterns. Learning the patterns of such interactions would enhance DTI accuracy if learning employs large, well-balanced datasets that cover all relationships between drugs and target proteins.
在药物研发中,识别最佳候选药物非常重要。生物学和计算科学领域的研究人员一直在寻求利用机器学习(ML)来有效预测药物-靶点相互作用(DTIs)。近年来,鉴于预训练模型在自然语言处理(NLPs)中日益凸显的作用,针对化合物和靶蛋白的预训练模型也在不断开发。本研究旨在使用针对化合物的基于变换器的双向编码器表征(BERT)预训练模型ChemBERTa来改进DTI预测模型。预训练采用简化分子输入线性条目系统(SMILES)。我们还将预训练的ProBERT用于靶蛋白(预训练采用氨基酸序列)。使用BIOSNAP、DAVIS和BindingDB数据库(单独或组合使用)进行学习。由ChemBERTa和ProtBert以及整合后的数据库共同训练的最终模型,与之前的模型相比,基于曲线下面积(AUC)和精确召回率-AUC值,展现出了迄今为止最佳的DTI预测性能。通过对13对底物与代谢酶细胞色素P450(CYP)的具体案例研究,验证了最终模型的性能。最终模型给出了出色的DTI预测结果。由于药物与靶蛋白之间的实际相互作用预计会呈现特定模式,使用ChemBERTa和ProtBert进行预训练可以传授这些模式。如果学习采用涵盖药物与靶蛋白之间所有关系的大规模、均衡数据集,那么了解此类相互作用的模式将提高DTI预测的准确性。