Department of Biostatistics, School of Public Health, Xuzhou Medical University, Xuzhou, 221004, Jiangsu, China.
Center for Medical Statistics and Data Analysis, Xuzhou Medical University, Xuzhou, 221004, Jiangsu, China.
BMC Bioinformatics. 2022 Aug 30;23(1):359. doi: 10.1186/s12859-022-04897-3.
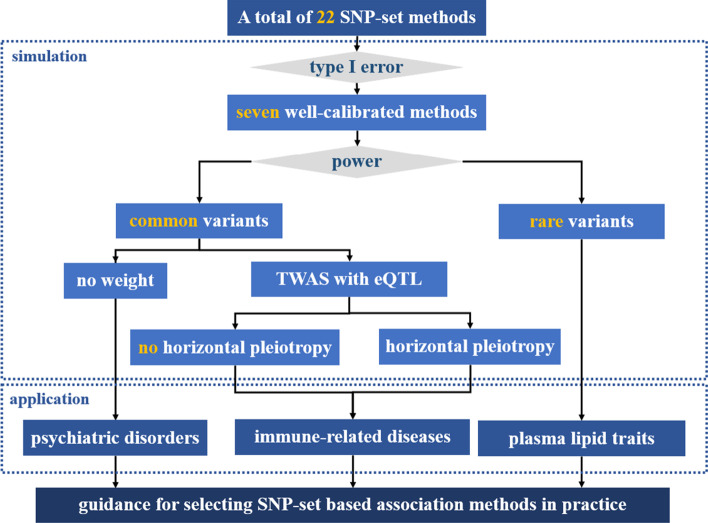
Multilocus analysis on a set of single nucleotide polymorphisms (SNPs) pre-assigned within a gene constitutes a valuable complement to single-marker analysis by aggregating data on complex traits in a biologically meaningful way. However, despite the existence of a wide variety of SNP-set methods, few comprehensive comparison studies have been previously performed to evaluate the effectiveness of these methods.
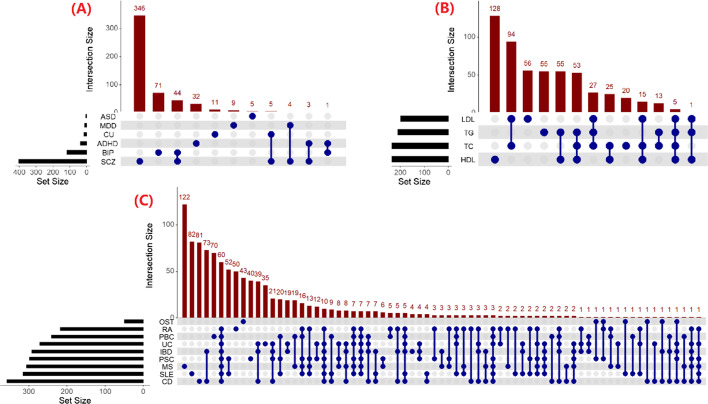
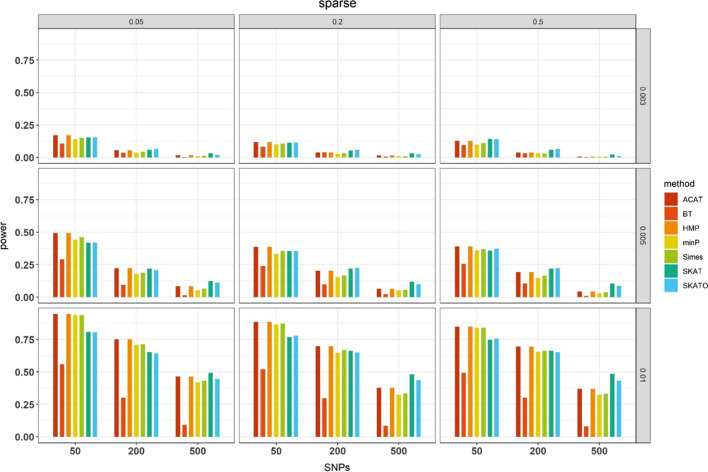
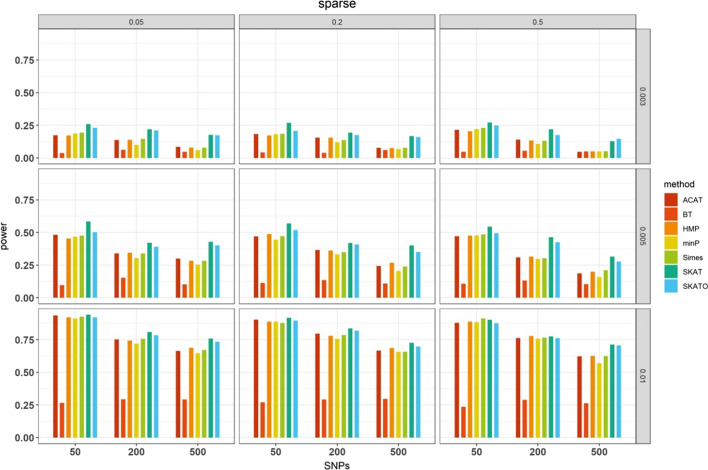
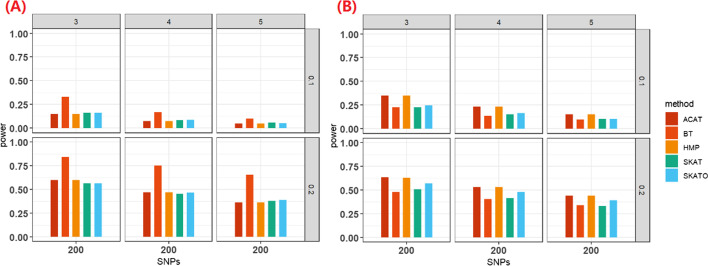
We herein sought to fill this knowledge gap by conducting a comprehensive empirical comparison for 22 commonly-used summary-statistics based SNP-set methods. We showed that only seven methods could effectively control the type I error, and that these well-calibrated approaches had varying power performance under the simulation scenarios. Overall, we confirmed that the burden test was generally underpowered and score-based variance component tests (e.g., sequence kernel association test) were much powerful under the polygenic genetic architecture in both common and rare variant association analyses. We further revealed that two linkage-disequilibrium-free P value combination methods (e.g., harmonic mean P value method and aggregated Cauchy association test) behaved very well under the sparse genetic architecture in simulations and real-data applications to common and rare variant association analyses as well as in expression quantitative trait loci weighted integrative analysis. We also assessed the scalability of these approaches by recording computational time and found that all these methods can be scalable to biobank-scale data although some might be relatively slow.
In conclusion, we hope that our findings can offer an important guidance on how to choose appropriate multilocus association analysis methods in post-GWAS era. All the SNP-set methods are implemented in the R package called MCA, which is freely available at https://github.com/biostatpzeng/ .
在基因内预先分配的一组单核苷酸多态性(SNP)的多位点分析通过以生物学上有意义的方式聚合复杂性状的数据,是对单标记分析的宝贵补充。然而,尽管存在各种各样的 SNP 集方法,但以前很少进行全面的比较研究来评估这些方法的有效性。
我们旨在通过对 22 种常用的基于汇总统计量的 SNP 集方法进行全面的实证比较来填补这一知识空白。我们表明,只有七种方法能够有效地控制 I 型错误,并且这些校准良好的方法在模拟场景下具有不同的功效表现。总体而言,我们证实了负担检验通常功效不足,基于分数的方差分量检验(例如序列核关联检验)在常见和罕见变异关联分析中的多基因遗传结构下具有更强的功效。我们进一步揭示了两种无连锁不平衡的 P 值组合方法(例如调和平均值 P 值方法和聚合柯西关联检验)在模拟和真实数据应用中常见和罕见变异关联分析以及表达数量性状基因座加权综合分析中的稀疏遗传结构下表现非常好。我们还通过记录计算时间来评估这些方法的可扩展性,发现尽管有些方法可能相对较慢,但所有这些方法都可以扩展到生物库规模的数据。
总之,我们希望我们的研究结果能够为在后 GWAS 时代如何选择适当的多基因关联分析方法提供重要指导。所有的 SNP 集方法都在名为 MCA 的 R 包中实现,可在 https://github.com/biostatpzeng/ 上免费获得。