Qi Jing, Sheng Qiongyu, Zhou Yang, Hua Jiao, Xiao Shutong, Jin Shuilin
School of Mathematics, Harbin Institute of Technology, Harbin, People's Republic of China.
Cell Biosci. 2022 Sep 2;12(1):142. doi: 10.1186/s13578-022-00886-4.
Single-cell RNA sequencing (scRNA-seq) provides a powerful tool to capture transcriptomes at single-cell resolution. However, dropout events distort the gene expression levels and underlying biological signals, misleading the downstream analysis of scRNA-seq data.
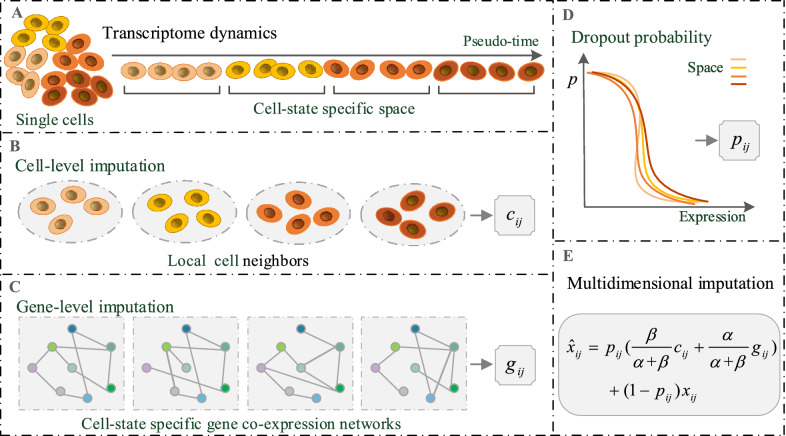
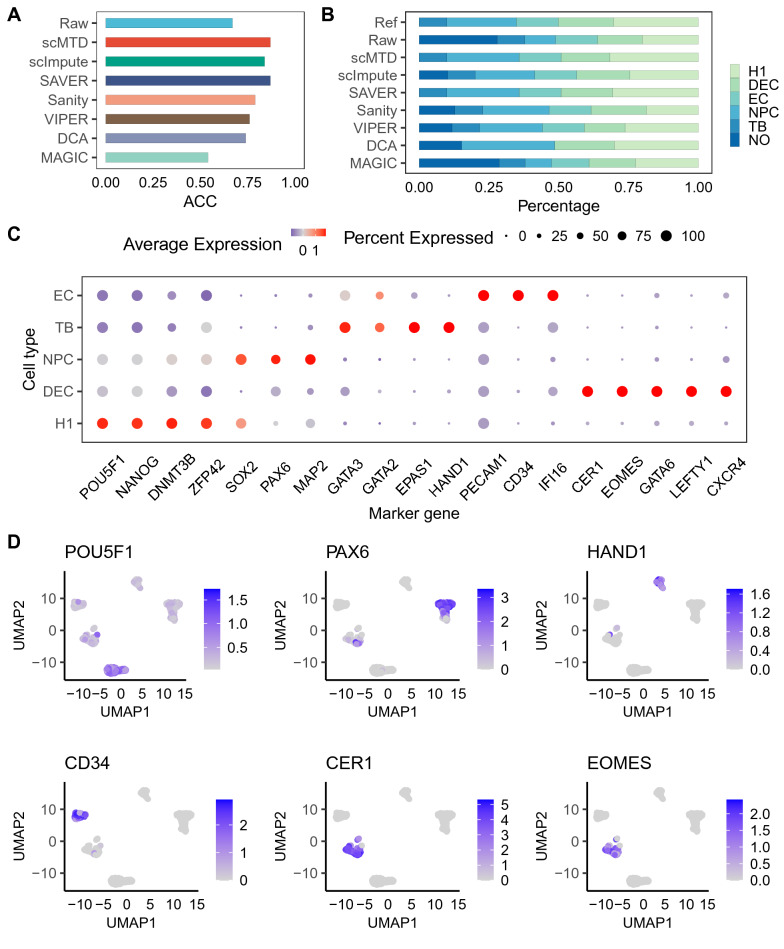
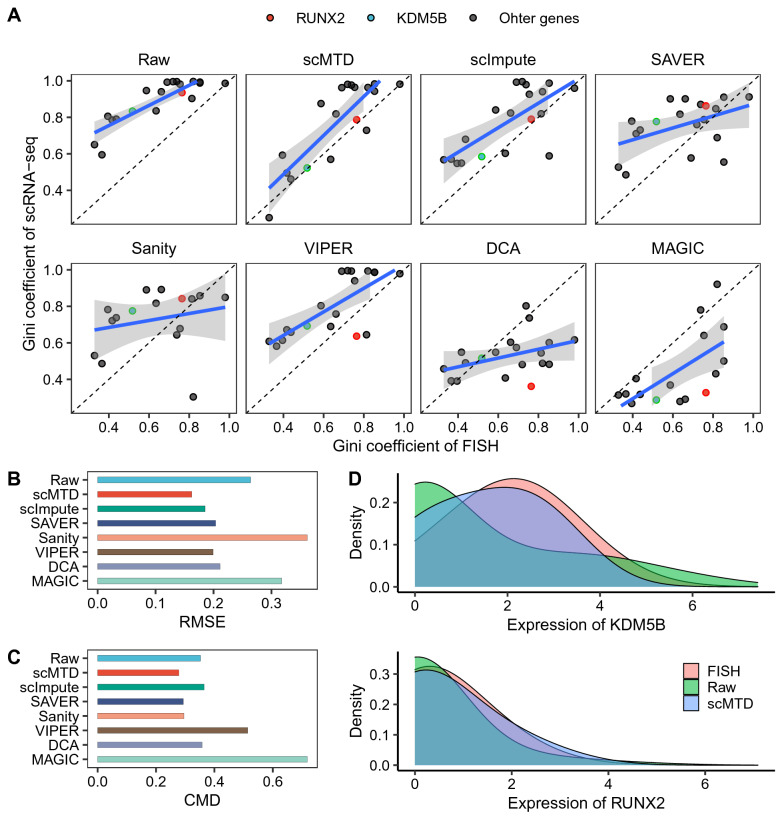
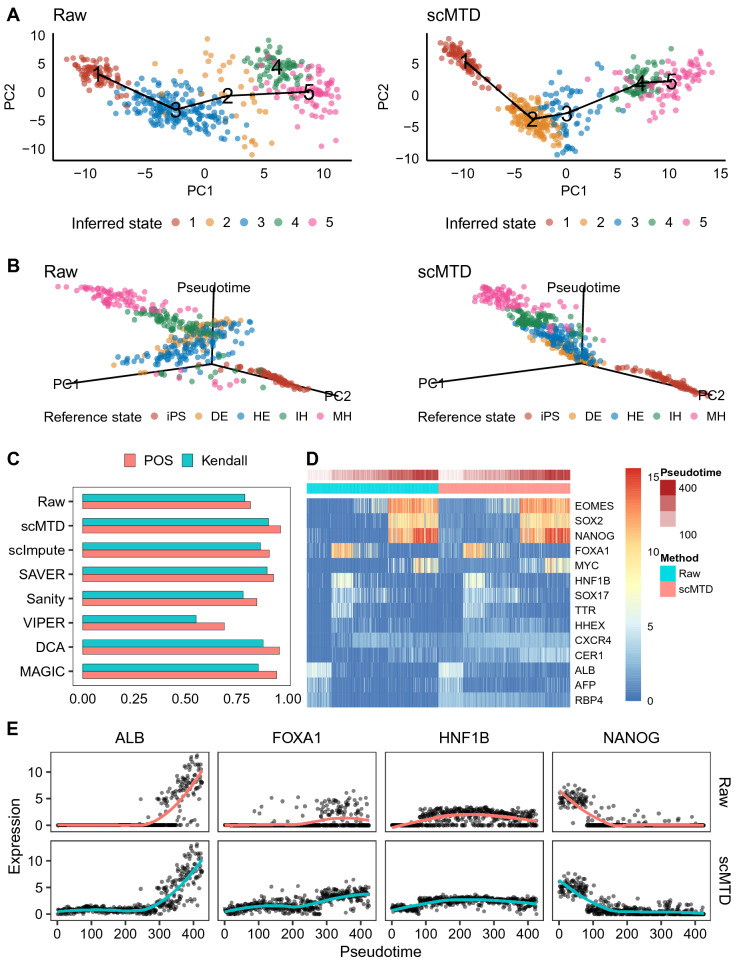
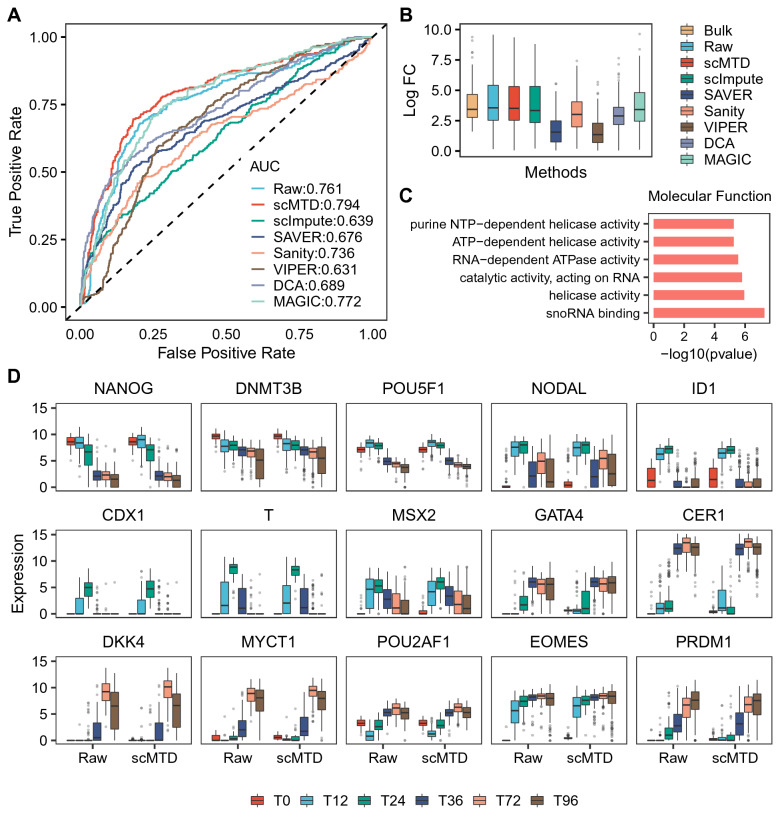
We develop a statistical model-based multidimensional imputation algorithm, scMTD, that identifies local cell neighbors and specific gene co-expression networks based on the pseudo-time of cells, leveraging information on cell-level, gene-level, and transcriptome dynamic to recover scRNA-seq data. Compared with the state-of-the-art imputation methods through several real-data-based analytical experiments, scMTD effectively recovers biological signals of transcriptomes and consistently outperforms the other algorithms in improving FISH validation, trajectory inference, differential expression analysis, clustering analysis, and identification of cell types.
scMTD maintains the gene expression characteristics, enhances the clustering of cell subpopulations, assists the study of gene expression dynamics, contributes to the discovery of rare cell types, and applies to both UMI-based and non-UMI-based data. Overall, scMTD's reliability, applicability, and scalability make it a promising imputation approach for scRNA-seq data.
单细胞RNA测序(scRNA-seq)为在单细胞分辨率下捕获转录组提供了一个强大的工具。然而,数据丢失事件会扭曲基因表达水平和潜在的生物学信号,误导scRNA-seq数据的下游分析。
我们开发了一种基于统计模型的多维插补算法scMTD,该算法基于细胞的伪时间识别局部细胞邻居和特定的基因共表达网络,利用细胞水平、基因水平和转录组动态信息来恢复scRNA-seq数据。通过几个基于真实数据的分析实验与最先进的插补方法相比,scMTD有效地恢复了转录组的生物学信号,并且在改善荧光原位杂交验证、轨迹推断、差异表达分析、聚类分析和细胞类型识别方面始终优于其他算法。
scMTD保持了基因表达特征,增强了细胞亚群的聚类,有助于基因表达动态的研究,有助于发现稀有细胞类型,并且适用于基于UMI和非基于UMI的数据。总体而言,scMTD的可靠性、适用性和可扩展性使其成为一种很有前景的scRNA-seq数据插补方法。